
Epitope-based Multi-variant SARS-Cov-2 Vaccine Design: Shared Epitopes Among the Natural SARS-Cov-2 Spike Glycoprotein and 5 of its Variants (D614G, α, β, γ, δ) with High in Silico Binding Affinity to Human Leukocyte Antigen (HLA) Class II Molecules
Spyros A. Charonis1,2, Apostolos P. Georgopoulos1,2*
1The HLA SARS-CoV-2 Research Group, Brain Sciences Center, Department of Veterans Affairs Health Care System, Minneapolis, MN 55417, USA
2Department of Neuroscience, University of Minnesota Medical School, Minneapolis, MN 55455, USA
Abstract and Introduction
The appearance and fast spread of five SARS-CoV-2 variants (D614G, B.1.1.7-UK [α], B.1.351-South Africa [β], P.1-Brazil [γ], B.167.2-India [δ]) have raised concerns regarding adaptive immunity, namely the extent to which antibodies against the original SARS-CoV-2 spike glycoprotein (Snatural) would protect against those variants1. A related issue is how effective current vaccines are against the known variants of concern2. This issue is important because all current vaccines have Snatural as their target antigen. The first step in initiating antibody production is the formation of a complex between an epitope of the foreign antigen (here, a spike glycoprotein) and a Human Leukocyte Antigen (HLA) Class II molecule; this complex engages CD4+ T-lymphocytes for the initiation of antibody production by B cells (Major Histocompatibility Complex [MHC] restriction)3-6. Given the underlying mechanisms of long-term adaptive immunity, vaccines containing epitopes shared by all 6 spike glycoprotein variants and with high binding affinity to HLA Class II molecules could potentially be good candidates for offering a universal protection against SARS-Cov-2. Numerous efforts have been made in determining vaccine targets for SARS-CoV-2 using computational methods. These methods are based on B cell and T cell epitope prediction7-14 and indeed extend beyond SARS-CoV-2 to encompass prediction of the pathogen-based immune response more generically15-19. In the current study, we explored this approach by investigating in silico the binding affinities of all linear 15-, 18- and 22-amino acid long epitopes of Snatural and its 5 variants (SD614G, SB.1.1.7, SB.1.351, SP.1, SP.167.2) to 66 common HLA Class II alleles with global frequencies of ≥ 0.01. We identified 18 such epitopes which occur in all 6 spike glycoproteins and which bind with very high affinity to HLA Class II molecules. Most of these molecules came from the DPB1 gene. The suitability of these candidate epitopes for a successful multivariant SARS-CoV-2 vaccine design remains to be determined.
Materials and Methods
The main objective of this study was to exhaustively assess the binding affinities of HLA Class II molecules to six variants of the SARS-CoV-2 spike glycoprotein. The variants and some of their sequence properties are summarized in Table 1. The point mutations and deletions of important SARS-CoV-2 variants are documented in several online repositories, with the CDC database (https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html) being used in this study.
HLA Alleles
For this study, we selected the more frequent alleles of classical HLA Class II genes (DPB1, DQB1, DRB1), namely all alleles with frequencies ≥ 0.01, an arbitrary but reasonable threshold. For that purpose, we obtained an Estimation of Global Allele Frequencies by querying the relevant website20. The alleles with frequencies ≥ 0.01 that we used are listed in Table 2. They comprised 21, 15 and 30 alleles of DPB1, DQB1 and DRB1 genes, respectively.
Partitioning the SARS-CoV-2 Spike Glycoprotein Variants
The amino acid sequences of five viral spike proteins (Snatural, SD614G, SB.1.1.7, SB.1.351, and S P.1) (Table 1) were retrieved from the UniprotKB database21. The amino acid sequence of the more recent Indian variant spike glycoprotein SP.167.2 was retrieved from the NCBI SARS-CoV-2 data hub22. The retrieved sequence (GenBank Acc: QWU05442.1) was matched by filtering the search for SARS-CoV-2 spike glycoprotein sequences from the B.167.2 lineage originating in India.
Each viral sequence was queried for binding affinity against 66 common HLA Class II alleles (Table 2). A sliding epitope window approach23 was used to partition the sequence of the spike glycoprotein for each variant. Partitioning was done in a manner to obtain all possible consecutive linear 15-, 18- and 22-mers (e.g. for 15-mers residues 1-15, 2-16, …, n-15 where n=sequence length) that cover the entire sequence length (Fig 1). These peptide lengths are in the range of suitable lengths for binding with HLA Class II molecules6.
The partitioning was implemented in a Python script (version 3.8). All n-mers were queried in the IEDB database24 in order to determine their binding affinities to a set of 66 HLA Class II receptor molecules. Binding affinity predictions were obtained using the NetMHCIIpan method25. For each n-mer, a binding affinity score was predicted and reported as a percentile rank by comparing the peptide’s score against the scores of five million random n-mers selected from the UniProt database21. Smaller percentile ranks indicate higher binding affinity. For each gene locus (e.g. DRB1) and spike protein variant (e.g. Indian delta variant), all alleles and n-mers (formatted as a FASTA sequence alignment) were entered as a single query and, thus, the same set of 5 million random n-mers was employed to rank all queried alleles. Altogether, for each allele, 7544 15-mers, 7526 18-mers, and 7502 22-mers were tested for a total of 22572 n-mers x 66 alleles = 1498464 tests. Smaller percentile ranks indicate higher binding affinity; therefore, the lowest (i.e. minimum) percentile rank (LPR) for each allele (corresponding to the highest binding affinity) and n-mer of each spike glycoprotein was retrieved. Finally, we employed the most conservative threshold of LPR = 0.01 (the lowest LPR returned by NetMHCIIpan) to identify selected epitope sequences (n-mers) with LPR = 0.01 (highest affinity) for further analysis.
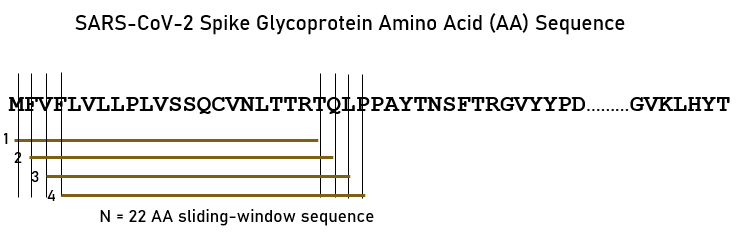
Figure 1.A sample of the sliding window approach23 for the spike glycoprotein variants (SARS-CoV-2 sequence displayed). See text for details.
Table 1:SARS-CoV-2 spike glycoprotein variants.
|
Variant/Location |
Nomenclature |
Length of Viral Protein |
N of 15-mers |
N of 18-mers |
N of 22-mers |
|
Natural |
SARS-CoV-2 |
1273 |
1258 |
1255 |
1251 |
|
D614G |
D614G / Asp614Gly |
1273 |
1258 |
1255 |
1251 |
|
UK (alpha, α) |
B.1.1.7 |
1271 |
1256 |
1253 |
1249 |
|
South Africa (beta, β) |
B.1.351 |
1273 |
1258 |
1255 |
1251 |
|
Brazil (gamma, γ) |
P.1 |
1273 |
1258 |
1255 |
1251 |
|
India (delta, δ) |
B.167.2 |
1271 |
1256 |
1253 |
1249 |
Table 2.HLA Class II alleles used, ordered by gene (color-coded) and their global frequencies in descending order. (See text for details.)
|
Index |
Allele |
Frequency |
|
1 |
DPB1*04:01 |
0.23022 |
|
2 |
DPB1*101:01 |
0.17700 |
|
3 |
DPB1*05:01 |
0.16296 |
|
4 |
DPB1*04:02 |
0.16181 |
|
5 |
DPB1*02:01 |
0.15451 |
|
6 |
DPB1*03:01 |
0.06760 |
|
7 |
DPB1*01:01 |
0.05857 |
|
8 |
DPB1*13:01 |
0.04415 |
|
9 |
DPB1*14:01 |
0.04115 |
|
10 |
DPB1*02:02 |
0.02725 |
|
11 |
DPB1*09:01 |
0.02473 |
|
12 |
DPB1*17:01 |
0.02403 |
|
13 |
DPB1*28:01 |
0.01827 |
|
14 |
DPB1*77:01 |
0.01597 |
|
15 |
DPB1*11:01 |
0.01460 |
|
16 |
DPB1*18:01 |
0.01448 |
|
17 |
DPB1*107:01 |
0.01400 |
|
18 |
DPB1*10:01 |
0.01295 |
|
19 |
DPB1*21:01 |
0.01172 |
|
20 |
DPB1*22:01 |
0.01149 |
|
21 |
DPB1*06:01 |
0.01001 |
|
22 |
DQB1*03:01 |
0.24528 |
|
23 |
DQB1*02:01 |
0.13308 |
|
24 |
DQB1*03:02 |
0.10029 |
|
25 |
DQB1*05:01 |
0.09970 |
|
26 |
DQB1*06:02 |
0.07696 |
|
27 |
DQB1*02:02 |
0.07162 |
|
28 |
DQB1*03:03 |
0.05454 |
|
29 |
DQB1*04:02 |
0.05185 |
|
30 |
DQB1*06:01 |
0.05043 |
|
31 |
DQB1*05:02 |
0.04806 |
|
32 |
DQB1*05:03 |
0.04270 |
|
33 |
DQB1*06:03 |
0.04042 |
|
34 |
DQB1*06:04 |
0.02836 |
|
35 |
DQB1*04:01 |
0.02426 |
|
36 |
DQB1*06:09 |
0.01356 |
|
37 |
DRB1*07:01 |
0.11305 |
|
38 |
DRB1*15:01 |
0.09560 |
|
39 |
DRB1*03:01 |
0.08850 |
|
40 |
DRB1*11:01 |
0.07516 |
|
41 |
DRB1*01:01 |
0.06829 |
|
42 |
DRB1*13:01 |
0.05585 |
|
43 |
DRB1*11:04 |
0.05065 |
|
44 |
DRB1*04:01 |
0.04420 |
|
45 |
DRB1*13:02 |
0.03842 |
|
46 |
DRB1*16:01 |
0.03479 |
|
47 |
DRB1*14:01 |
0.03005 |
|
48 |
DRB1*14:54 |
0.02665 |
|
49 |
DRB1*15:02 |
0.02583 |
|
50 |
DRB1*12:01 |
0.02435 |
|
51 |
DRB1*04:04 |
0.02248 |
|
52 |
DRB1*09:01 |
0.02224 |
|
53 |
DRB1*04:05 |
0.02144 |
|
54 |
DRB1*08:01 |
0.02097 |
|
55 |
DRB1*12:02 |
0.02014 |
|
56 |
DRB1*04:03 |
0.01807 |
|
57 |
DRB1*01:02 |
0.01745 |
|
58 |
DRB1*13:03 |
0.01670 |
|
59 |
DRB1*04:11 |
0.01642 |
|
60 |
DRB1*08:03 |
0.01629 |
|
61 |
DRB1*04:07 |
0.01524 |
|
62 |
DRB1*16:02 |
0.01494 |
|
63 |
DRB1*14:02 |
0.01480 |
|
64 |
DRB1*10:01 |
0.01429 |
|
65 |
DRB1*08:02 |
0.01421 |
|
66 |
DRB1*04:02 |
0.01362 |
The locations in the primary sequence of all 15-, 18- and 22-mers with LPR = 0.01 were tabulated and the n-mers that overlapped with the receptor-binding domain (RBD) of the spike glycoprotein identified. This sequence-based quantity was calculated as the number of residues within the RBD (positions 338-506 in the linear sequence of the spike protein) divided by the length of the respective n-mer, i.e., 15/18/22. RBD proportion values were included because RBD is the structural region that binds to ACE2 receptors26. Indeed, serological studies of over 600 individuals infected with SARS-CoV-2 have shown that ~90% of the plasma or serum-neutralizing antibody activity targets the spike protein RBD27.
Results
We identified 18 epitope sequences which occurred in all 6 spike glycoproteins and had HLA binding affinities of LPR = 0.01 for at least one allele. Table 3 shows the AA sequence of each epitope and their position in the relevant glycoprotein sequence. Table 4 shows the alleles for which a sequence had very high in silico binding affinity of LPR = 0.01, together with the sum of the population frequencies of those alleles as an estimate of global population coverage; this estimate will vary among different populations, depending on the allele frequency specific for a particular population. Finally, the fraction of overlap of each sequence with the RBD region is given in Table 5. Remarkably, all 4 sequences with most high binding affinities (#2, 3, 4, 12, in bold in Table 4) highly overlapped with the RBD region.
Table 3.Position of the 18 epitopes for Snatural and its 5 variants in Table 1.
|
|
SARS-CoV-2 Variant |
|||||||
|
Epitope |
n-mer |
AA Sequence |
Natural |
D614G |
α |
β |
γ |
δ |
|
1 |
15 |
DEMIAQYTSALLAGT |
867 |
867 |
864 |
866 |
867 |
865 |
|
2 |
15 |
FGEVFNATRFASVYA |
338 |
338 |
336 |
338 |
338 |
336 |
|
3 |
15 |
GEVFNATRFASVYAW |
339 |
339 |
337 |
339 |
339 |
337 |
|
4 |
15 |
PFGEVFNATRFASVY |
337 |
337 |
335 |
337 |
337 |
335 |
|
5 |
15 |
QQLIRAAEIRASANL |
1010 |
1010 |
1007 |
1009 |
1010 |
1008 |
|
6 |
15 |
TDEMIAQYTSALLAG |
866 |
866 |
863 |
865 |
866 |
864 |
|
7 |
15 |
TQQLIRAAEIRASAN |
1009 |
1009 |
1006 |
1008 |
1009 |
1007 |
|
8 |
18 |
FGEVFNATRFASVYAWNR |
338 |
338 |
336 |
338 |
338 |
336 |
|
9 |
18 |
GEVFNATRFASVYAWNRK |
339 |
339 |
337 |
339 |
339 |
336 |
|
10 |
18 |
LLTDEMIAQYTSALLAGT |
864 |
864 |
861 |
863 |
864 |
336 |
|
11 |
18 |
LTDEMIAQYTSALLAGTI |
865 |
865 |
862 |
864 |
865 |
337 |
|
12 |
18 |
PFGEVFNATRFASVYAWN |
337 |
337 |
335 |
337 |
337 |
337 |
|
13 |
18 |
TYVTQQLIRAAEIRASAN |
1006 |
1006 |
1003 |
1005 |
1006 |
863 |
|
14 |
22 |
ITNLCPFGEVFNATRFASVYAW |
332 |
332 |
330 |
332 |
332 |
330 |
|
15 |
22 |
TNLCPFGEVFNATRFASVYAWN |
333 |
333 |
331 |
333 |
333 |
331 |
|
16 |
22 |
NLCPFGEVFNATRFASVYAWNR |
334 |
334 |
332 |
334 |
334 |
332 |
|
17 |
22 |
LCPFGEVFNATRFASVYAWNRK |
335 |
335 |
333 |
335 |
335 |
333 |
|
18 |
22 |
CPFGEVFNATRFASVYAWNRKR |
336 |
336 |
334 |
336 |
336 |
334 |
Table 4. HLA alleles for which a sequence had a very high in silico binding affinity of LPR = 0.01. The cumulative allele frequency is the sum of the global frequencies of the corresponding alleles, given in Table 2. The sequences with higher population coverage are in bold.
|
Epitope |
n-mer |
AA Sequence |
Allele number (from Table 2) |
Cumulative allele frequency |
|
1 |
15 |
DEMIAQYTSALLAGT |
38, 49 |
0.1214 |
|
2 |
15 |
FGEVFNATRFASVYA |
1, 2, 4, 7, 10, 13, 14, 20 |
0.7006 |
|
3 |
15 |
GEVFNATRFASVYAW |
1, 2, 4, 7, 10, 14, 20 |
0.5053 |
|
4 |
15 |
PFGEVFNATRFASVY |
1, 4, 7, 14 |
0.4666 |
|
5 |
15 |
QQLIRAAEIRASANL |
26, 30 |
0.1274 |
|
6 |
15 |
TDEMIAQYTSALLAG |
38, 49 |
0.1214 |
|
7 |
15 |
TQQLIRAAEIRASAN |
26, 30 |
0.1274 |
|
8 |
18 |
FGEVFNATRFASVYAWNR |
1, 7, 20 |
0.3003 |
|
9 |
18 |
GEVFNATRFASVYAWNRK |
20 |
0.0115 |
|
10 |
18 |
LLTDEMIAQYTSALLAGT |
49 |
0.0258 |
|
11 |
18 |
LTDEMIAQYTSALLAGTI |
49 |
0.0258 |
|
12 |
18 |
PFGEVFNATRFASVYAWN |
1, 4, 7, 14 |
0.4781 |
|
13 |
18 |
TYVTQQLIRAAEIRASAN |
30 |
0.0504 |
|
14 |
22 |
ITNLCPFGEVFNATRFASVYAW |
1 |
0.2302 |
|
15 |
22 |
TNLCPFGEVFNATRFASVYAWN |
1 |
0.2302 |
|
16 |
22 |
NLCPFGEVFNATRFASVYAWNR |
1 |
0.2302 |
|
17 |
22 |
LCPFGEVFNATRFASVYAWNRK |
1 |
0.2302 |
|
18 |
22 |
CPFGEVFNATRFASVYAWNRKR |
1 |
0.2302 |
Table 5. RBD overlap of the 18 epitopes for Snatural and its 5 variants in Table 1.
|
|
SARS-CoV-2 Variant |
|||||||
|
Epitope |
n-mer |
AA Sequence |
Natural |
D614G |
α |
β |
γ |
δ |
|
1 |
15 |
DEMIAQYTSALLAGT |
0 |
0 |
0 |
0 |
0 |
0 |
|
2 |
15 |
FGEVFNATRFASVYA |
0.93 |
0.93 |
0.80 |
0.93 |
0.93 |
0.80 |
|
3 |
15 |
GEVFNATRFASVYAW |
1.00 |
1.00 |
0.87 |
1.00 |
1.00 |
0.87 |
|
4 |
15 |
PFGEVFNATRFASVY |
0.87 |
0.87 |
0.73 |
0.87 |
0.87 |
0.73 |
|
5 |
15 |
QQLIRAAEIRASANL |
0 |
0 |
0 |
0 |
0 |
0 |
|
6 |
15 |
TDEMIAQYTSALLAG |
0 |
0 |
0 |
0 |
0 |
0 |
|
7 |
15 |
TQQLIRAAEIRASAN |
0 |
0 |
0 |
0 |
0 |
0 |
|
8 |
18 |
FGEVFNATRFASVYAWNR |
0.94 |
0.94 |
0.83 |
0.94 |
0.94 |
0.83 |
|
9 |
18 |
GEVFNATRFASVYAWNRK |
1.00 |
1.00 |
0.89 |
1.00 |
1.00 |
0.83 |
|
10 |
18 |
LLTDEMIAQYTSALLAGT |
0 |
0 |
0 |
0 |
0 |
0.83 |
|
11 |
18 |
LTDEMIAQYTSALLAGTI |
0 |
0 |
0 |
0 |
0 |
0.89 |
|
12 |
18 |
PFGEVFNATRFASVYAWN |
0.89 |
0 |
0.78 |
0.89 |
0.89 |
0.89 |
|
13 |
18 |
TYVTQQLIRAAEIRASAN |
0 |
0 |
0 |
0 |
0 |
0 |
|
14 |
22 |
ITNLCPFGEVFNATRFASVYAW |
0.68 |
0.68 |
0.59 |
0.68 |
0.68 |
0.59 |
|
15 |
22 |
TNLCPFGEVFNATRFASVYAWN |
0.73 |
0.73 |
0.64 |
0.73 |
0.73 |
0.64 |
|
16 |
22 |
NLCPFGEVFNATRFASVYAWNR |
0.77 |
0.77 |
0.68 |
0.77 |
0.77 |
0.68 |
|
17 |
22 |
LCPFGEVFNATRFASVYAWNRK |
0.82 |
0.82 |
0.73 |
0.82 |
0.82 |
0.73 |
|
18 |
22 |
CPFGEVFNATRFASVYAWNRKR |
0.86 |
0.86 |
0.77 |
0.86 |
0.86 |
0.77 |
Alleles Involved
Of the total 66 alleles tested (Table 2), 21 belonged to DPB1 gene, 15 to DQB1 gene, and 30 to DRB1 gene. On the other hand, there were 12 distinct alleles (Table 6) involved in high binding affinity with the 18 sequences identified. Most of them (8/12; 66.7%) belonged to the DPB1 gene, with only 2/12 coming from the DQB1 and DRB1 genes, each. With respect to the number of alleles tested, again the highest proportion of high affinity binding alleles came from the DPB1 gene (8/21; 38.1%), followed by the DQB1 gene (2/15; 13.3%) and the DRB1 gene (2/30; 6.7%).
Table 6.HLA alleles involved in very high in silico binding affinity of LPR = 0.01 with the 18 sequences.
|
Allele |
Number of sequences involved |
Percent of 18 sequences in which allele was involved |
|
DPB1*01:01 |
5 |
27.8% |
|
DPB1*02:02 |
2 |
11.1 |
|
DPB1*04:01 |
10 |
55.6 |
|
DPB1*04:02 |
4 |
22.2 |
|
DPB1*22:01 |
4 |
22.2 |
|
DPB1*28:01 |
1 |
5.6 |
|
DPB1*77:01 |
4 |
22.2 |
|
DPB1*101:01 |
2 |
11.1 |
|
DQB1*06:01 |
3 |
16.7 |
|
DQB1*06:02 |
2 |
11.1 |
|
DRB1*15:01 |
2 |
11.1 |
|
DRB1*15:02 |
4 |
22.2 |
Discussion
The rationale of this study rests on the HLA restriction on antigen presentation to CD4+ T lymphocytes. More specifically, the formation of HLA Class II molecule-peptide antigen complex is a primary, necessary, although not sufficient, stage in successful antibody production, which depends on other factors downstream from CD4+ T cell activation. Here we focused on exhaustively screening in silico the affinity of linear continuous epitopes of the natural SARS-CoV-2 spike glycoprotein and of 5 of its variants (D614G, B.1.1.7, B.1.1351, P.1, P167.2) to common 66 HLA Class II molecules (global frequency ≥ 0.01) in an effort to identify epitopes occurring in all 6 spike glycoproteins that bind with very high affinity of HLA Class II molecules. We reasoned that such epitopes would be good candidates for vaccine(s) that would be effective against all 6 SARS-CoV-2 spike glycoproteins. Indeed, we identified 18 such epitopes that occur in all 6 glycoproteins and bind with very high affinity (LPR = 0.01) with various Class II alleles (Table 5). These epitopes pass the first screening for high affinity binding with HLA Class II molecules and, pending, further assessment28-29, are suitable candidates for being employed in a SARS-CoV-2 multivariant vaccine design. In addition, Table 4 provides estimates of global population coverage for each sequence with 4 sequences (#2, 3, 4, 12 in Table 4) offering high coverage; estimates of population coverage would vary for different populations, depending on the frequencies of the alleles involved in a particular population. Remarkably, the position of all those 4 sequences in each glycoprotein greatly overlaps with the RBD region.
Finally, with respect to the HLA Class II genes involved in this high affinity binding to epitopes of the 6 SARS-CoV-2 spike glycoproteins, the DPB1 gene provided the highest percentages of alleles involved (Table 6). In contrast, both the DQB1 and DRB1 genes were much less involved. The prominent involvement of the DPB1 gene is in keeping with our previous finding that the frequency of alleles of the DPB1 gene is positively associated with the binding affinity of epitopes of the SARS-CoV-2 natural spike glycoprotein23. This may reflect an evolutionary pressure favoring the selection of the DPB1 gene due to its presumed success in binding to coronaviruses, thus aiding survival and conferring evolutionary advantage.
Limitations
The main limitation of these results is that they were derived using an in silico methodology. We believe that this approach is justified given the infeasibility of performing those binding affinity assessments experimentally.
Acknowledgments
Partial funding for this study was provided by the University of Minnesota (the Brain and Genomics Fund and the American Legion Brain Sciences Chair). The sponsors had no role in the current study design, analysis or interpretation, or in the writing of this paper. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Author contributions
APG conceived the research; SAC carried out the viral protein retrieval, IEDB query and data preprocessing; APG contributed to data analysis; SAC and APG contributed to writing the manuscript.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Lauring AS, Hodcroft EB. Genetic variants of SARS-CoV-2-What do they mean? JAMA. 2021; 325: 529-531.
- Williams TC, Burgers WA. SARS-CoV-2 evolution and vaccines: cause for concern? Lancet Respir. Med. 2021; Jan 29: S2213-2600(21)00075-8.
- Swain SL. T cell subsets and the recognition of MHC class. Immunological Rev. 1983; 74: 129-142.
- Rudensky AY, Preston-Hurlburt P, Hong SC, et al. Sequence analysis of peptides bound to MHC Class II molecules. Nature. 1991; 353: 622-627.
- Weenink SM, Gautam AM. Antigen presentation by MHC class II molecules. Immunol. Cell Biol. 1997; 75: 69-81.
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu. Rev. Immunol. 2013; 31: 443-473.
- Ahmed SF, Quadeer AA, McKay MR. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses. 2020 Feb 25;12(3): 254. doi: 10.3390/v12030254. PMID: 32106567; PMCID: PMC7150947.
- Arwa A. Mohammed, Shaza W, et al. "Epitope-Based Peptide Vaccine against Glycoprotein G of Nipah Henipavirus Using Immunoinformatics Approaches", Journal of Immunology Research, vol. 2020, Article ID 2567957, 12 pages, 2020. https://doi.org/10.1155/2020/2567957
- Grifoni A, Sidney J, Zhang Y, et al. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe. 2020 Apr 8;27(4):671-680.e2. doi: 10.1016/j.chom.2020.03.002. Epub 2020 Mar 16. PMID: 32183941; PMCID: PMC7142693.
- Kiyotani K, Toyoshima Y, Nemoto K. et al. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J Hum Genet 65, 569–575 (2020). https://doi.org/10.1038/s10038-020-0771-5.
- Lon JR, Bai Y, Zhong B, et al. Prediction and evolution of B cell epitopes of surface protein in SARS-CoV-2. Virol J 17, 165 (2020). https://doi.org/10.1186/s12985-020-01437-4
- Poran A, Harjanto D, Malloy M, et al. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med 12, 70 (2020). https://doi.org/10.1186/s13073-020-00767-w.
- Renu Jakhar, S. K. Gakhar. "An Immunoinformatics Study to Predict Epitopes in the Envelope Protein of SARS-CoV-2", Canadian Journal of Infectious Diseases and Medical Microbiology, vol. 2020, Article ID 7079356, 14 pages, 2020. https://doi.org/10.1155/2020/7079356
- Phan IQ, Subramanian S, Kim D. et al. In silico detection of SARS-CoV-2 specific B-cell epitopes and validation in ELISA for serological diagnosis of COVID-19. Sci Rep 11, 4290 (2021). https://doi.org/10.1038/s41598-021-83730-y
- Paul S, Lindestam Arlehamn CS, Scriba TJ, et al. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 2015 Jul; 422: 28-34. doi: 10.1016/j.jim.2015.03.022. Epub 2015 Apr 7. PMID: 25862607; PMCID: PMC4458426.
- Paul S, Sidney J, Sette A, et al. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr Protoc Immunol. 2016; 114: 18.19.1-18.19.24. Published 2016 Aug 1. doi:10.1002/cpim.12.
- Ali MT, Morshed MM, Hassan F. A Computational Approach for Designing a Universal Epitope-Based Peptide Vaccine Against Nipah Virus. Interdiscip Sci. 2015 Jun; 7(2): 177-85. doi: 10.1007/s12539-015-0023-0. Epub 2015 Jul 9. PMID: 26156209.
- Khan MA, Hossain MU, Rakib-Uz-Zaman SM, et al. Epitope-based peptide vaccine design and target site depiction against Ebola viruses: an immunoinformatics study. Scand J Immunol. 2015 Jul; 82(1): 25-34. doi: 10.1111/sji.12302. PMID: 25857850; PMCID: PMC7169600.
- Oyarzun P, Ellis JJ, Gonzalez-Galarza FF, et al. A bioinformatics tool for epitope-based vaccine design that accounts for human ethnic diversity: application to emerging infectious diseases. Vaccine. 2015 Mar 3; 33(10): 1267-73. doi: 10.1016/j.vaccine.2015.01.040. Epub 2015 Jan 25. PMID: 25629524.
- The Allele Frequency Database, http://www.allelefrequencies.net/
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47, D506-D515.
- NCBI SARS-CoV-2 data hub, www.ncbi.nlm.nih.gov/sars-cov-2.
- Charonis SS, Tsilibary EP, Georgopoulos AP. SARS-CoV-2 virus and Human Leukocyte Antigen (HLA) class II: Investigation in silico of binding affinities for COVID-19 protection and vaccine development. J. Immunological Sci. 2020; 4(4): 12-23.
- Immune Epitope Database and Analysis Resource, www.iedb.org.
- Reynisson B, Alvarez B, Paul S, et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020; 48, W449-W454.
- Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020 May; 581(7807): 215-220. doi: 10.1038/s41586-020-2180-5. Epub 2020 Mar 30. PMID: 32225176.
- Piccoli L, Park YJ, Tortorici MA, et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell. 2020 Nov 12; 183(4): 1024-1042.e21. doi: 10.1016/j.cell.2020.09.037. Epub 2020 Sep 16. PMID: 32991844; PMCID: PMC7494283.
- Flower DR. Designing immunogenic peptides. Nat Chem Biol. 2013; 9(12):749-753.
- 29. Van Regenmortel MHV. Antigenicity and immunogenicity of synthetic peptides. Biologicals. 2001; 29: 209-213.