
Intestinal bacterial biofilms modulate mucosal immune responses
Melissa Ellermann1 and R. Balfour Sartor2, 3, 4*
1Department of Microbiology, University of Texas Southwestern Medical Center, Dallas, TX, USA.
2Departments of Medicine, University of North Carolina, Chapel Hill, NC, USA.
3Microbiology and Immunology, University of North Carolina, Chapel Hill, NC, USA.
4Center for Gastrointestinal Biology and Disease, University of North Carolina, Chapel Hill, NC, USA.
Abstract
Host-associated microbial communities modulate numerous aspects of host physiology at the epithelial interface within mucosal environments. Perturbations to this symbiotic relationship between host and microbiota has been linked to numerous microbial-driven pathological states, including Crohn’s disease (CD). This is in part driven by the outgrowth of aggressive resident bacterial strains such as adherent and invasive Escherichia coli (AIEC) and changes in bacterial physiology and function that promote enhanced mucosal association of pathobionts and aberrant stimulation of mucosal immunity. Endogenous bacteria from host-associated microbial communities can adopt a sessile lifestyle and form multicellular structures known as biofilms that are generated through the expression of extracellular adhesion factors that include curli amyloid fibrils, cellulose and type 1 pili. In addition to enabling bacterial attachment to mucosal surfaces, biofilm components also stimulate immune responses and can therefore instigate or perpetuate microbial-driven inflammatory diseases such as CD. These host-bacterial interactions provide pharmacological targets that can potentially be exploited to limit mucosal adherence of aggressive enteric bacteria, inappropriate stimulation of inflammatory immune responses and consequent development of chronic intestinal inflammation.
Host-associated microbial communities, termed microbiota, have been implicated in the pathogenesis of Crohn’s disease (CD)1. Community-wide profiling of the intestinal microbiota in humans and in rodent models have correlated characteristic compositional and functional changes to the microbiota with various pathological states. In addition to decreased bacterial diversity, one common feature of the disease-associated microbiota in CD is the outgrowth of more aggressive members of intestinal bacteria, known as pathobionts that instigate or perpetuate effector immune responses in susceptible hosts. Gnotobiotics, in conjunction with microbial and mouse genetic tools, have further enabled the identification of specific functional host-microbial interactions that play a causative role in initiating or perpetuating the development of intestinal inflammation and may serve as promising therapeutic targets for mitigating inflammation in CD.
The GI tract is home to a complex collection of distinct microbial communities that are subject to modulation by biogeographical factors. Intestinal microbiota are physically separated from the underlying mucosal immune system by the mucosal barrier that is composed of a single layer of epithelial cells, a thick layer of mucus and host secretions including antimicrobial peptides and soluble IgA antibodies recognizing luminal microbial antigens. Proximity to the mucus layer is one biogeographical factor that compositionally and functionally shapes microbial communities. Indeed, the composition and transcriptional profiles of the luminal and mucosal microbiota are distinct2, 3. In the non-inflamed intestinal environment, resident bacteria form biofilm-like structures within the outer mucus layer that are comprised of bacterial aggregates encased within extracellular matrix components2. In disease states such as inflammatory bowel diseases (IBD) or colorectal cancer (CRC), these mucus-associated bacterial aggregates can penetrate the inner mucus layer and adhere to the epithelial interface4, 5, thus enabling more direct stimulation of epithelial and immune cells. This close proximity provides the opportunity for components of in vivo biofilms within the GI tract to modulate mucosal immunity.
The spatial organization of bacterial biofilms and the environmental cues that stimulate biofilm formation has largely been studied in vitro with monocultures on abiotic substrates. As a result, in vivo biofilm formation and function, particularly in the GI tract, are not well understood. Nonetheless, in vitro models have been instrumental in defining the bacterial-derived components that constitute biofilms. Recent work has also demonstrated the immunogenicity of various extracellular matrix (ECM) components present within bacterial biofilms and ECM modulation of in vivo bacterial fitness, localization and interactions with the host.
Enterobacteriaceae
In CD, the mucus-associated microbiota undergoes profound compositional alterations (dysbiosis) that is in part characterized by decreased bacterial diversity that includes expansion of Enterobacteriaceae, including CD-associated Escherichia coli known as adherent and invasive E. coli (AIEC)6, 7, 8. AIEC are functionally characterized by their ability to invade epithelial cells, survive and replicate within epithelial cells and macrophages and induce colitis and bacterial- specific T cell responses in murine models of chronic intestinal inflammation9, 10, 11, 12, 13. AIEC also form robust biofilms and express components often present within Enterobacteriaceae biofilms, including curli, cellulose, type 1 pili, flagellin and extracellular DNA9, 14, 15, 16, 17, 18. These biofilm components have been implicated in modulating the inflammatory potential of AIEC and other resident and pathogenic members of the Enterobacteriaceae family in the contexts of infectious and pathobiont-driven colitis. This review summarizes the mechanisms by which three ECM components, curli fibrils, cellulose and type 1 pili, interact with the mucosal immune system to drive or exacerbate the pathogenesis of microbial-driven inflammatory diseases such as CD, with a specific focus on Enterobacteriaceae (Figure 1). The modulation of mucosal immunity by flagellin and bacterial DNA (inflammasome activation) in the context of IBD has been reviewed elsewhere19, 20, 21.
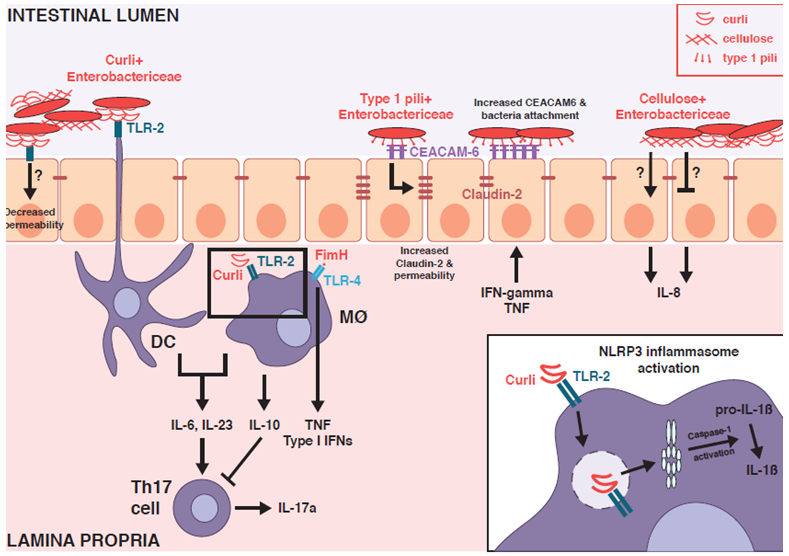
Figure 1: Mucosal immune stimulation by Enterobacteriaceae biofilm components.
Curli fibrils, type 1 pili and cellulose - common extracellular matrix components present within Enterobacteriaceae biofilms - stimulate mucosal immune responses and modulate epithelial barrier function through engagement with Toll-like receptors (TLRs), NOD-like receptors (NLRs) and carcinoembryonic antigen-related cell adhesium molecules (CEACAMs). This provides putative mechanisms by which biofilm components can impact colitis activity in the inflamed intestinal environment within susceptible hosts.
Curli fibrils
Amyloid fibrils are synthesized within biofilms formed by various bacterial phyla present within the intestinal microbiota including Bacteroidetes, Firmicutes and Proteobacteria22, 23. Curli are the most well studied bacterial amyloid and are produced by Enterobacteriaceae including resident intestinal E. coli and invading enteric pathogens such as Salmonella24, 25. Early evidence demonstrating the immunogenicity of curli was reported in one study where anti-curli antibodies were recovered from sepsis patients with blood cultures positive for curli-expressing E. coli26. More recent work has demonstrated that curli is also recognized by the innate immune system. Curli serves as a ligand for the Toll-like receptor (TLR) heterodimer TLR1/2 and stimulates immune responses in variety of cell types27. Curli also activates the NOD-like receptor protein 3 (NLRP3) inflammasome and induces caspase-1 activation and maturation of pro-interleukin-(IL)-1β28. Additionally, human amyloids promote the development of neurodegenerative diseases such as Alzheimer’s and serve as danger associated molecular patterns (DAMPs) that interact with TLRs expressed by microglia29. These findings introduce the possibility that amyloids synthesized by other bacterial members of the microbiota may also modulate immune responses and host-microbial interactions at mucosal interfaces and thus warrants further investigation.
While the contribution of curli to the inflammatory potential of AIEC has not been directly investigated, infectious colitis models demonstrate the importance of curli as a modulator of epithelial barrier function and mucosal immunity in the intestines. One study showed that infection with curli-expressing Salmonella enhanced T-helper-(Th)-17 immune responses in a TLR-2 dependent manner while not impacting luminal pathogen fitness30. Ex vivo studies further demonstrated that dendritic cells (DC) were specifically responsive to purified curli fibrils30, suggesting that DC sampling of curli may promote a tissue microenvironment conducive to Th17 activation. Curli directly reinforces epithelial barrier function through TLR-2 signaling by reducing epithelial permeability and bacterial translocation and enhancing epithelial secretion of IL-831. Finally, oral administration of purified curli confers protection against chemically-induced colitis by stimulating mucosal expression of the anti-inflammatory cytokine IL-10 through a TLR-2 dependent mechanism32. Taken together, these studies collectively demonstrate that curli serves as a protective bacterial factor that limits infectious and chemically-induced colitis.
The effects of curli on immune activation are context dependent. In contrast to the effects of oral administration of purified curli, intraperitoneal injection of curli had no effect on splenic IL-10 expression and instead enhanced splenic interferon gamma (IFN-γ) production32. Within Salmonella biofilms, curli forms tight complexes with extracellular DNA; these complexes synergistically activate type I IFN responses in innate immune cells33, 34. Intraperitoneal administration of either purified curli-DNA complexes or curli-expressing Salmonella or resident E. coli promoted expression of type I IFNs and production of autoreactive antibodies against dsDNA and chromatin in systemic lupus erythematosus-(SLE)-resistant wild type and SLE-susceptible mice33. The curli-DNA complex is endocytosed by innate immune cells through interactions with TLR-2, which subsequently enables TLR-9 stimulation by curli-bound DNA within the endosome34. This in turn stimulates the production of type I IFNs that correspond with enhanced autoimmunity34. Given the contrasting effects of curli in different animal models, it will be interesting to investigate whether curli produced by resident bacteria modulates mucosal immunity in a protective or pathological manner in chronic colitis models and in IBD.
Bacterial cellulose
Cellulose is an exopolysaccharide and biofilm ECM component produced by numerous bacterial taxa including Enterobacteriaceae35. Demonstrating direct evidence of bacterial cellulose production within in vivo environments such as the GI tract has been challenging because bacterial cellulose production is post-transcriptionally regulated35. Therefore, in contrast to other biofilm components, correlating the expression of cellulose biosynthetic genes and cellulose biosynthesis is not a meaningful indicator of ongoing cellulose production. An added complication for detecting enteric bacteria cellulose in vivo is that it is chemically indistinguishable from dietary plant cellulose. Thus, most studies to date have relied on generating cellulose-deficient bacterial mutants to define the contribution of cellulose production to bacterial fitness and interactions with the host. Interestingly, cellulose production at the physiological temperature of 37°C is more common in fecal than uropathogenic E. coli isolates25. Moreover, environmental stimuli present in the intestines, such as iron availability, peroxide stress and IgA monoclonal antibodies, stimulate in vitro cellulose production by Salmonella and E. coli36, 37, 16. Cellulose biosynthesis by phagolysosomal Salmonella has also been demonstrated in macrophages in vitro38. In aggregate, these findings provide compelling evidence that resident intestinal E. coli and invading enteric pathogens synthesize cellulose within the intestinal environment.
Direct stimulation of immune responses by bacterial cellulose has not been demonstrated. However, bacterial genetic studies provide evidence that bacterial cellulose production modulates host-microbial interactions and disease induction in AIEC-driven colitis. Abrogation of cellulose production in the intestinal probiotic E. coli strain Nissle reduces in vitro epithelial adherence and induction of IL-8, possibly suggesting that cellulose may be required to induce maximal invasion and consequent inflammatory responses39. In contrast, in a different fecal E. coli isolate, cellulose counteracts curli-mediated enhancement of epithelial adhesion and IL-8 induction40. Cellulose-mediated modulation of macrophage inflammatory responses also varies with the same AIEC strain and is dependent on iron availability16. In the intestinal environment, cellulose production accelerated colitis induction by AIEC in inflammation-susceptible, mono-colonized Il10-/- mice, corresponded with enhanced luminal fitness and augmented pathological Th17 mucosal immunity16. However, the precise mechanisms by which AIEC cellulose production enhances colitis induction in Il10-/- mice remain to be elucidated. Collectively, these findings support a model where cellulose likely modulates host immunity through an indirect mechanism that is characterized by cellulose fibers masking other immunogenic components expressed on the bacterial cell surface.
Type 1 pili
Type 1 pili are extracellular appendages often expressed by biofilm-associated Enterobacteriaceae that mediate bacterial attachment to host cells and contribute to biofilm formation41, 15. Type 1 pili are comprised of FimA protein subunits that form the pilus and the FimH adhesin localized at the end of each pilus that binds mannose residues on receptors expressed by host cells41. In addition to enabling direct bacterial interactions with host cells, FimH stimulates innate immune responses including inflammatory cytokine secretion and neutrophil transepithelial migration through its interactions with TLR-442, 43.
Type 1 pili are an established microbiota-derived factor that enhances bacterial fitness and exacerbates inflammation in AIEC-driven models of colitis. Carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) serves as a receptor for type 1 pili and is expressed on the apical side of intestinal epithelial cells44. Inflammatory stimuli augment expression of CEACAM644, thus promoting the establishment of a tissue microenvironment that favors AIEC epithelial colonization. Indeed, transgenic expression of human CEACAM6 in CEABAC10 mice enables colonic tissue colonization of AIEC in a FimH-dependent manner45. In contrast to their wild type counterparts, CEABAC10 mice challenged with AIEC exhibit epithelial barrier dysfunction and develop severe colitis that is attenuated by blockade of CEACAM6 or colonization with a FimH-deficient AIEC mutant45, 46. The inflamed intestinal environment itself promotes increased AIEC expression of fimH47, thus further perpetuating this dysbiotic host-pathobiont interaction. Targeting FimH with antagonists that inhibit interactions with CEACAM6 reduces intestinal colonization of AIEC and ameliorates colitis induction in CEABAC10 mice48. Taken together, these studies demonstrate how specific interactions between bacterial biofilm components and host cells promote the development of intestinal inflammation and can be pharmacologically targeted to limit dysbiotic host-pathobiont interactions and consequent colitis induction.
Summary and future directions
While the focus of this review is on Enterobacteriaceae, it is important to emphasize that the bacterial biodiversity of the mucus-associated bacterial niche along the intestinal tract is high and comprises members of many additional bacterial taxa and mycobiome species4, 2, 8, 5, 49, 50. Accordingly, changes in abundance of bacterial and fungal taxa in addition to Enterobacteriaceae have also been correlated with the pathogenesis of chronic, microbial-driven diseases such as CD and CRC4, 8, 5, 49, 50. Indeed, adhesins from other bacteria such as Fusobacterium nucleatum modulate epithelial barrier function and mucosal immunity to promote a carcinogenic microenvironment51, 52, 53. Biofilm components also stimulate protective immune responses. For example, ECM components from Fecalibacterium prausnitzii biofilms ameliorate the severity of chemically induced colitis54. Collectively, these studies demonstrate that biofilm components from diverse bacterial taxa can modulate mucosal immune responses in the GI tract. Understanding the precise interactions between in vivo bacterial biofilms and the host could lead to the identification of novel pharmacological targets that can potentially be exploited to limit mucosal adherence of pathobionts, minimize effector immune responses and augment protective mucosal immunity in individuals susceptible for developing microbial-driven inflammatory disorders such as IBD.
Acknowledgements
This work was supported by grants to RB Sartor from the Crohn’s and Colitis Foundation of America (Microbiome Consortium), NIH P01 DK094779, NIH P40 OD01995, NIH P30 DK34987 and NIH R01 DK053347.
Conflict of interest
The authors declare no conflict of interest.
References
- Sartor RB, Wu GD. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology. 2017; 152: 327–339.e4.
- Macfarlane S, Bahrami B, Macfarlane GT. Mucosal biofilm communities in the human intestinal tract. Adv. Appl. Microbiol. 2011; 75: 111–143.
- Li H, Limenitakis JP, Fuhrer T, et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nature Communications. 2015; 6: 8292.
- Swidsinski A, Weber J, Loening-Baucke V, et al. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005; 43: 3380–3389.
- Dejea CM, Wick EC, Hechenbleikner EM, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proceedings of the National Academy of Sciences. 2014; 111: 18321–18326.
- Martin HM, Campbell BJ, Hart CA, et al. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004; 127: 80–93.
- Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. 2004; 127: 412–421.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014; 15: 382–392.
- Boudeau J, Barnich N, Darfeuille-Michaud A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn's disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol. 2001; 39: 1272–1284.
- Glasser AL, Boudeau J, Barnich N, et al. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001; 69: 5529–5537.
- Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005; 128: 891–906.
- Small CLN, Reid-Yu SA, McPhee JB, et al. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nature Communications. 2013; 4: 1957.
- Eun CS, Mishima Y, Wohlgemuth S, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect Immun. 2014; 82: 2239–2246.
- Martinez-Medina M, Naves P, Blanco J, et al. Biofilm formation as a novel phenotypic feature of adherent-invasive Escherichia coli (AIEC). BMC Microbiol. 2009; 9: 202.
- Hung C, Zhou Y, Pinkner JS, et al. Escherichia coli biofilms have an organized and complex extracellular matrix structure. mBio. 2013; 4: e00645–13.
- Ellermann M, Huh EY, Liu B, et al. Adherent-Invasive Escherichia coli Production of Cellulose Influences Iron-Induced Bacterial Aggregation, Phagocytosis, and Induction of Colitis. Infect Immun. 2015; 83: 4068–4080.
- Carvalho FA, Koren O, Goodrich JK, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012; 12: 139–152.
- la Fuente De M, Franchi L, Araya D, et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int J Med Microbiol. 2014; 304: 384–392.
- Elia PP, Tolentino YFM, Bernardazzi C, et al. The role of innate immunity receptors in the pathogenesis of inflammatory bowel disease. Mediators of Inflammation. 2015; 2015: 936193–10.
- Vijay-Kumar M, Gewirtz AT. Flagellin: key target of mucosal innate immunity. Mucosal Immunology. 2009; 2: 197–205.
- Chen GY, Núñez G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology. 2011; 141: 1986–1999.
- Larsen P, Nielsen JL, Dueholm MS, et al. Amyloid adhesins are abundant in natural biofilms. Environmental Microbiology. 2007; 9: 3077–3090.
- Jordal PB, Dueholm MS, Larsen P, et al. Widespread abundance of functional bacterial amyloid in mycolata and other gram-positive bacteria. Appl Environ Microbiol. 2009; 75: 4101–4110.
- Römling U, Sierralta WD, Eriksson K, et al. Multicellular and aggregative behaviour of Salmonella typhimurium strains is controlled by mutations in the agfD promoter. Mol Microbiol. 1998; 28: 249–264.
- Bokranz W, Wang X, Tschäpe H, et al. Expression of cellulose and curli fimbriae by Escherichia coli isolated from the gastrointestinal tract. Journal of Medical Microbiology. 2005; 54: 1171–1182.
- Bian Z, Brauner A, Li Y, et al. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J. Infect. Dis. 2000; 181: 602–612.
- Tükel Ç, Nishimori JH, Wilson RP, et al. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol. 2010; 12: 1495–1505.
- Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, et al. Toll-Like Receptor 2 and NLRP3 Cooperate To Recognize a Functional Bacterial Amyloid. Curli Infect Immun. 2015; 83: 693–701.
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nature Reviews Immunology. 2014; 14: 463–477.
- Nishimori JH, Newman TN, Oppong GO, et al. Microbial Amyloids Induce Interleukin 17A (IL-17A) and IL-22 Responses via Toll-Like Receptor 2 Activation in the Intestinal Mucosa. Infect Immun. 2012; 80: 4398–4408.
- Oppong GO, Rapsinski GJ, Newman TN, et al. Epithelial Cells Augment Barrier Function via Activation of the Toll-Like Receptor 2/Phosphatidylinositol 3-Kinase Pathway upon Recognition of Salmonella enterica Serovar Typhimurium Curli Fibrils in the Gut. Infect Immun. 2013; 81: 478–486.
- Oppong GO, Rapsinski GJ, Tursi SA, et al. Biofilm-associated bacterial amyloids dampen inflammation in the gut: oral treatment with curli fibres reduces the severity of hapten-induced colitis in mice. NPJ Biofilms and Microbiomes. 2015; 1: 804.
- Gallo PM, Rapsinski GJ, Wilson RP, et al. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity. 2015; 42: 1171–1184.
- Tursi SA, Lee EY, Medeiros NJ, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. 2017; 13: e1006315.
- Römling U, Galperin MY. Bacterial cellulose biosynthesis: diversity of operons, subunits, products, and functions. Trends in Microbiology. 2015; 23: 545–557.
- Depas WH, Hufnagel DA, Lee JS, et al. Iron induces bimodal population development by Escherichia coli. Proceedings of the National Academy of Sciences. 2013; 110: 2629–2634.
- Amarasinghe JJ, D'Hondt RE, Waters CM, et al. Exposure of Salmonella enterica Serovar typhimurium to a protective monoclonal IgA triggers exopolysaccharide production via a diguanylate cyclase-dependent pathway. Infect Immun. 2013; 81: 653–664.
- Pontes MH, Lee EJ, Choi J, et al. Salmonella promotes virulence by repressing cellulose production. Proceedings of the National Academy of Sciences. 2015; 112: 5183–5188.
- Monteiro C, Saxena I, Wang X, et al. Characterization of cellulose production in Escherichia coli Nissle 1917 and its biological consequences. Environmental Microbiology. 2009; 11: 1105–1116.
- Wang X, Rochon M, Lamprokostopoulou A, et al. Impact of biofilm matrix components on interaction of commensal Escherichia coli with the gastrointestinal cell line HT-29. CMLS. Cell Mol Life Sci. 2006; 63: 2352–2363.
- Bouguénec CL. Adhesins and invasins of pathogenic Escherichia coli. International Journal of Medical Microbiology. 2005; 471–478. doi:10.1016/j.ijmm.2005.07.001
- Mossman KL, Mian MF, Lauzon NM, et al. Cutting Edge: FimH Adhesin of Type 1 Fimbriae Is a Novel TLR4 Ligand. The Journal of Immunology. 2008; 181: 6702–6706.
- Ashkar AA, Mossman KL, Coombes BK, et al. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathog. 2008; 4: e1000233.
- Barnich N, Carvalho FA, Glasser AL, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007; 117: 1566–1574.
- Carvalho FA, Barnich N, Sivignon A, et al. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009; 206: 2179–2189.
- Denizot J, Sivignon A, Barreau F, et al. Adherent-invasive Escherichia coli induce claudin-2 expression and barrier defect in CEABAC10 mice and Crohn's disease patients. Inflamm Bowel Dis. 2012; 18: 294–304.
- Arthur JC, Gharaibeh RZ, Mühlbauer M, et al. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nature Communications. 2014; 5: 4724.
- Sivignon A, Yan X, Alvarez Dorta D, et al. Development of Heptylmannoside-Based Glycoconjugate Antiadhesive Compounds against Adherent-Invasive Escherichia coli Bacteria Associated with Crohn's Disease. mBio. 2015; 6: e01298–15.
- Iliev ID, Funari VA, Taylor KD, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012; 336: 1314–1317.
- Hager CL, Ghannoum MA. The mycobiome: Role in health and disease, and as a potential probiotic target in gastrointestinal disease. Dig Liver Dis. 2017; 49: 1171–1176.
- Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013; 14: 195–206.
- Gur C, Ibrahim Y, Isaacson B, et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity. 2015; 42: 344–355.
- Fulbright LE, Ellermann M, Arthur JC. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017; 13: e1006480.
- Rossi O, Khan MT, Schwarzer M, et al. Faecalibacterium prausnitzii Strain HTF-F and Its Extracellular Polymeric Matrix Attenuate Clinical Parameters in DSS-Induced Colitis. PLoS ONE. 2015; 10: e0123013.