
A Flow Cytometric Immunophenotyping Approach to the Detection of Regulated Cell Death Processes
A Vossenkamper1, G Warnes2*
1Centre for Immunobiology, The Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary London University, 4 Newark Street, London E1 2AT, UK
2Flow Cytometry Core Facility, The Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary London University, 4 Newark Street, London E1 2AT, UK
Abstract
The use of the Western Blot technique has been the gold standard to determine protein expression and to semi-quantitate this expression in cell lysates. The recent publication of a flow cytometric immunophenotyping method employing fluorescently labelled antibodies to the intracellular labelling of antigens involved in Regulated Cell Death (RCD) processes has allowed the detection of three of these processes simultaneously which gave clarity to the interpretation of the relationship between apoptosis, RIP1 dependent apoptosis and necroptosis. Flow cytometry can now immunophenotype necroptosis by virtue of the up-regulation of RIP3 with simultaneous estimations of the degree of classic apoptosis (Caspase-3+ve/RIP3-ve) and of RIP1-dependent apoptosis (Caspase-3+ve/RIP3+ve) in live and dead cell populations. This approach for detecting multiple forms of cell death has been confirmed by the use of apoptosis and necroptosis blocking agents, zVAD and necrostatin-1 after treatment with etoposide or shikonin which induced apoptosis and necroptosis. The addition of anti-PARP and H2AX antibodies for the detection of parthanatos and DNA damage showed that double negative Caspase-3-ve/RIP3-ve cells detected in a previous study have undergone parthanatos or still display a negative phenotype for any cell death process.
The use of Western blot technology to quantitate types and levels of the various forms of regulated cell death (RCD) has been in place for decades. Although there are limitations with the technique1,2 there has been great advances in the understanding of the signalling pathways of apoptosis, necroptosis, RIP1-dependent apoptosis, pyroptosis, parthanatos and DNA damage and how they interact within the cell and there locations within the cells3-8. One of the main criticisms of the Western Blot is the number of cells required and hence that low incidence populations will not be detected. Also all the cells are lysed and hence there is no distinguishing of live and dead cells which may show dramatically different distribution of particular RCDs. Hence the relative levels of these different types of cell populations may unduly influence the quantification of specific forms of RCDs1. The use of fluorescent antibodies targeted against active Caspase-3 (apoptosis), RIP3 (necroptosis), PARP (parthanatos) and H2AX (DNA damage) and a fixable cell viability allows flow cytometric analysis of such cells which removes all the issues that have questioned the value of Western Blot analysis in cell death research. Moreover, this approach gives an insight into the relative incidences of most RCDs within a single cell population revealing that as perhaps expected live cells show a very different phenotypic profile to dead cells which are very uniform no matter which RCD has been induced9.
Materials and methods
Jurkat cells (human acute T cell leukaemia cell line) were left untreated or treated shikonin (S, 0.5 μM) or etoposide (Etop, 1 μM) for 24 h. Cells were also pre-treated with necroptosis blocker necrostatin-1 (60 μM) and or pan-caspase blocker zVAD (20 μM) for 2 h before shikonin or etoposide treatment.
Harvested cells were labelled with fixable live dead stain, Zombie NIR (Near Infra-Red) at RT for 15 mins. Cells were then washed in PBS/BSA and pellets fixed in Solution A (CalTag) for 15 min at RT. Washed cells were permeabilised in 0.25% Triton X-100 for 15 min at RT. Washed cells (0.5 x 106) were then labelled anti-RIP3-PE and anti- active caspase-3-BV650 for 20 min at RT. Washed cells were resuspended in 400 μl PBS and analysed on a ACEA Bioscience Novocyte 3000 flow cytometer (100,000 events). SEM calculated for percentage positive data and RIP3 MFI (n=3) Statistics for comparisons of treated samples to untreated cells only, Student t test P>0.05 not significant (NS), P<0.05*, P<0.01**, P<0.001***.
Polychromatic Flow Cytometric Analysis of RCD
The employment of fluorescently tagged antibodies that bind to proteins within intact cells as opposed to cell lysates has led the development of a flow cytometric immunophenotyping methodology to quantitate multiple forms of RCD simultaneously within live and dead cell populations within a single sample9. This was achieved by the availability of such antibodies that bind to the main defining proteins (e.g. active Caspase-3, RIP3, PARP and H2AX) usually employed in Western Blots. Hence classic apoptosis was defined by the use of active-casapse-3-BV650 in combination with a fixable live cell probe such as BioLegend’s Zombie NIR. This permitted the defining of cells undergoing early apoptosis compared to untreated Jurkat cells (Caspase-3+ve/cell viability-ve) as well as those cells that have died via apoptosis (Caspase-3+ve/cell viability+ve) or necrosis (Caspase-3-ve/cell viability±ve Figure. 1A, B)10-12. Shikonin a known inducer of necroptosis13 and apoptosis14 likewise showed a similar profile to that induced by the apoptosis-inducing drug etoposide (Figure. 1C). Blockade of etoposide induced apoptosis by pre-treatment with pan-caspase blocker zVAD resulted in a 50 % reduction of early apoptosis (Caspase-3+ve/cell viability-ve) with a small reduction in overall cell death (Figure. 1D)15. Similarly zVAD reduced detectable shikonin induced apoptosis by 60% with more cells dying via a caspase-3 independent mechanism commonly termed necrosis (Figure. 1E)13,15. Blockade of shikonin induced necroptosis by necrostatin-1 (Necro-1) caused a 40% increase in apoptosis with 38% less necrotic cell death (Caspase-3-ve/cell viability+ve Figure. 1F)13,15.
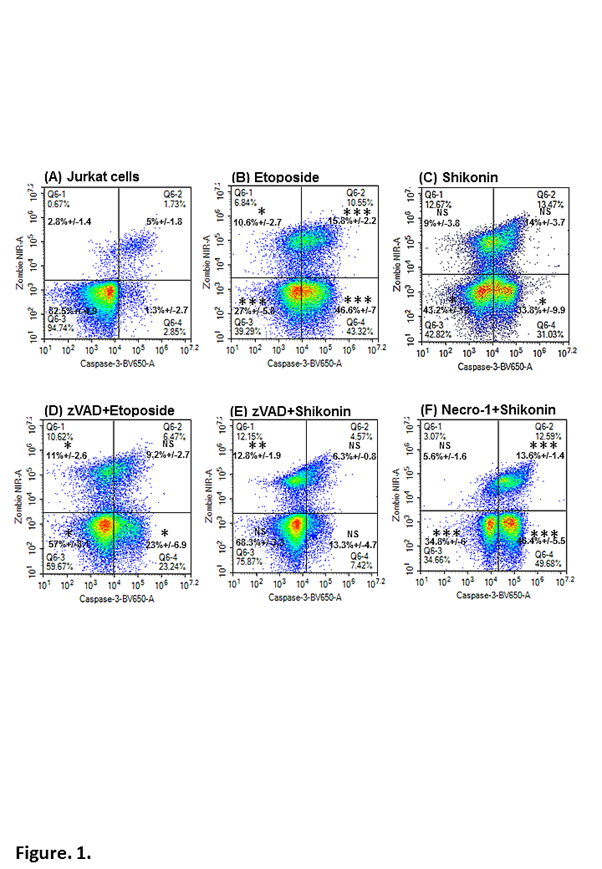
Figure 1: Flow cytometric detection of early and late apoptosis and necrotic cell death.
Jurkat cells were incubated with or without zVAD (20 μM), or necrostatin-1 (60 μM) with etoposide (1 μM), or shikonin (0.5 μM) for 24 h. Cells were labelled with the cell viability probe, Zombie NIR; fixed and permeabilised and then labelled with anti-active-caspase-3-BV-650. Lower left quadrant (Zombie NIR-ve/Caspase-3-ve) indicate live cells, early apoptosis (lower right quadrant) by Zombie NIR-ve/Caspase-3+ve, late apoptosis (upper right quadrant) by Zombie NIR+ve/Caspase-3+ve and necrotic cells (upper left quadrant) by Zombie NIR+ve/Caspase-3-ve. Student T-test for comparisons of treated and untreated cells P<0.05 *, <0.01 **, <0.001 *** NS –not significant, n=3.
However, a major stumbling block to the process was to phenotypically define necroptosis which occurs when RIP1 up-regulates RIP36,9,13,16. After trying 3 clones of anti-RIP3 antibody a clone was found that can detect this up-regulation of RIP3 in live cells (Caspase-3-ve/cell viability-ve) which included early apoptotic (Caspase-3+ve/cell viability-ve) and dead cells (Caspase-3+ve/cell viability+ve and Caspase-3-ve/cell viability+ve) using specific cell fixing and permeabilization reagents (Figure. 2). This then allowed the defining of apoptosis (Caspase-3+ve/RIP3-ve), resting (Caspase-3-ve/RIP3+ve) or necroptosis (Caspase-3-ve/RIP3+ve high) and RIP1-dependent apoptosis (Caspase-3+ve/RIP3+ve) within live and dead cell populations as defined by cell viability and caspase-3 (Figure. 2A, B). Live cells undergoing apoptosis showed a high level of the classic apoptotic phenotype (Caspase-3+ve/RIP3-ve) as well as RIP1-dependent apoptosis (Figure. 2C). RIP3 is associated with RIP1, therefore Caspase-3+ve/RIP3+ve events are classed as undergoing RIP1-dependent apoptosis (Figure. 2C). Dead cells produced during etoposide induced apoptosis reflect the situation in live cells with an equal amount of classic and RIP1-dependent apoptosis as shown in untreated dead cells (Figure. 2B, D). Blockade of etoposide-induced apoptosis by zVAD results in little classic apoptosis (<10%) in the live cell population (Figure. 2E). However the remaining Caspase-3+ve/cell viability-ve events shown in Figure. 1D are shown to be mainly undergoing RIP1-dependent apoptosis to a similar degree as that shown in etoposide treatment (Figure. 2E). A higher degree of live cells are Caspase-3-ve/RIP3+ve and double negative Caspase-3-ve/RIP3-ve (Figure. 2E). Dead cells from zVAD blockade have undergone RIP1-dependent apoptosis with as expected little classic apoptosis but the majority of cells have undergone death with a double negative phenotype (Figure. 2F).
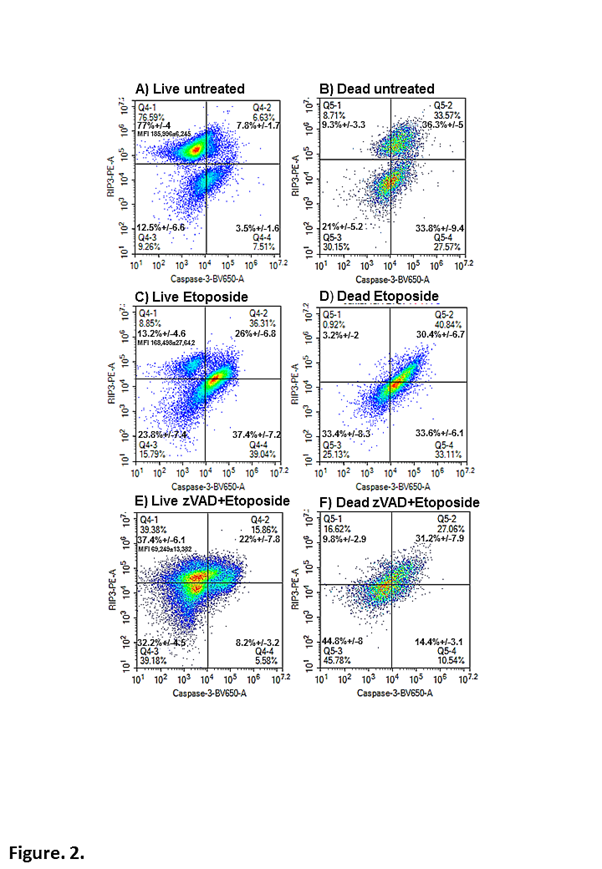
Figure 2: Immunophenotyping of apoptosis and RIP1-dependent apoptosis in live and dead cells.
Jurkat cell were untreated (A, B) or treated with etoposide (1 μM) (C, D) or with zVAD (20 μM) for 24 h (E, F). Cells were labelled with the cell viability probe, Zombie NIR, fixed and permeabilised and then labelled with RIP3-PE and anti-active-caspase-3-BV-650. After gating on live/early apoptotic and all dead cells as in Figure 1, apoptosis was indicated by Caspase-3+ve/RIP3-ve (lower right quadrant) and RIP1-dependent apoptosis Caspase-3+ve/RIP3+ve (upper right quadrant). While necroptosis was indicated by the up-regulation of RIP3 MFI (Median Fluorescence Intensity) in the upper left quadrant, Caspase-3-ve/RIP3high+ve.
Necroptosis was defined by the up-regulation of RIP3 (Median Fluorescence Intensity, MFI 215,524 compared to untreated Jurkat cells (naïve peripheral blood cells have a similar RIP3 expression, unpublished observation) 186,000 or a 16% up-regulation, NS) in the Caspase-3-ve/RIP3+ve population (32%) in the live cells (Figure. 3A)6,13,16. These live cells also showed a high degree of apoptosis (18%), RIP1-dependent apoptosis (26%) and a double negative phenotype (24%, Figure. 3A). Dead cells displayed a different distribution of apoptotic (33%), RIP1-dependent apoptosis (31%) and double negative phenotype (31%) compared to live cells (Figure. 3B)13,14. The defining of the live necroptotic population (Caspase-3-ve/RIP3+ve) by the up-regulation of RIP3 was confirmed by the blockade of shikonin induced necroptosis by necrostatin-1, which showed a down-regulation of RIP3 by >60% (Figure. 3C)13. Shikonin-induced apoptosis was enhanced due to blockade of the necroptosis pathway by necrostatin-1 while a similar level of RIP1-dependent apoptosis still went ahead (Figure. 1F, 3C). These cells also then went on to undergo cell death via apoptosis and RIP1-dependent apoptosis (Figure. 3D). Interestingly, when shikonin induced apoptosis was blocked by zVAD such live cells were either of the necroptotic phenotype (Caspase-3-ve/RIP3+ve) or double negative with no significant degree of RIP1-dependent apoptosis unlike that observed with etoposide (Figure 3E). Hence etoposide induced RIP1-dependent apoptosis appears to circumvent the mode of action of zVAD while shikonin did not, thus giving an insight into the signalling routes activated by the two drugs. However dead cells resulting from zVAD blockade of shikonin again showed a high degree of RIP1-dependent apoptosis (and double negative population) thus apparently circumventing this blockade of caspase-3 (Figure. 3F).
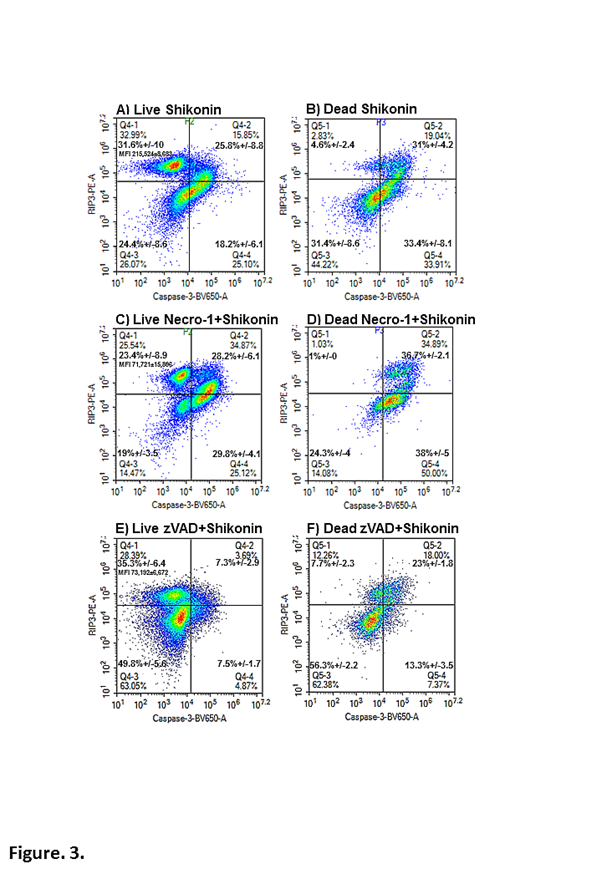
Figure 3: Immunophenotyping of necroptosis, apoptosis and RIP1-dependent apoptosis in live and dead cells. Jurkat cell were treated with shikonin (0.5 μM) (A, B) and with zVAD (20 μM) (C, D) or necrostatin-1 (60 μM) (Necro-1) (E, F) for 24 h. Cells were labelled with the cell viability probe, Zombie NIR; fixed and permeabilised and then labelled with RIP3-PE and anti-active-caspase-3-BV-650. After gating on live/early apoptotic and all dead cells as in Figure 1, apoptosis was indicated by Caspase-3+ve/RIP3-ve (lower right quadrant) and RIP1-dependent apoptosis Caspase-3+ve/RIP3+ve (upper right quadrant). While necroptosis was indicated by the up-regulation of RIP3 MFI (Median Fluorescence Intensity) in the upper left quadrant, Caspase-3-ve/RIP3high+ve.
Enhancement of the Assay
The increased presence of live and dead cells with a double negative phenotype after use of zVAD and necrostatin-1 raised the question how were these cells dying? Is it via pyroptosis, autophagy, parthanatos or DNA damage? So following on from the recent publication of this method it has been possible to include the detection of parthanatos and DNA damage by the use of PARP and H2AX antibodies extending the assay to five colour immunophenotyping.
We gated on the Caspase-3-ve/RIP3-ve double negative population Caspase-3-ve/RIP3-ve in live and dead cells and analysed the degree of parthanatos and DNA damage in untreated cells (PARP+ve/H2AX-ve or quadruple negative or Caspase-3-ve/RIP3-ve/PARP+ve/H2AX-ve, see Figure. 4A, B). Cells undergoing apoptosis (etoposide) saw a change in the level of expression of PARP and H2AX in the double negative population, in that more cells were PARP+ve/H2AX-ve and there was a 3 fold increase in the incidence of the PARP+ve/H2AX+ve population (Figure. 4C, D). After zVAD blockade of apoptosis the increase in the PARP+ve/H2AX-ve live cell population was maintained but the PARP+ve/H2AX+ve population was reduced compared to etoposide (Figure. 4E). This increase in live cell parthanatos was interestingly not transferred to the dead cell population which showed similar levels as that detected in dead untreated cells (Figure. 4B, F). Hence zVAD appears to abrogate the DNA damage response but not live cell parthanatos. Interestingly, co-treatment with necrostatin-1 and etoposide resulted in the abrogation of parthanatos in live and dead cells but displayed an increase in DNA damage resulting in a higher proportion of quadruple phenotypically live negative cells (Figure. 4G, H). The dual blockade of apoptosis by zVAD and necrostatin-1 resulted in the return of the parthanatos response in live cells as wells as double positive events (PARP+ve/H2AX+ve) which resulted in a reduction of the incidence of the quadruple negative phenotype (Figure. 4I). After a dual blockade of apoptosis and necroptosis, the dead cells showed a similar distribution of cells the same as that induced by etoposide (Figure. 4J).
Similar analysis of shikonin treated cells showed a high degree of live and dead cell parthanatos (PARP+ve/H2AX-ve) and a double positive dead cell population (PARP+ve/H2AX+ve, Figure. 4K, L). Shikonin with zVAD (like etoposide with zVAD) showed an increase in live and dead cell parthanatos (PARP+ve/H2AX-ve) but a decrease in the live double positive population to untreated levels (PARP+ve/H2AX+ve, Figure. 4M, N). Like necrostatin-1 blockade of apoptosis, live shikonin (with necrostatin-1) treated cells showed a similar increase in the double positive population with a decrease in parthanatos to control levels Figure. 4O). While like necrostatin-1 blockade of apoptosis, shikonin with necrostatain-1 then showed a doubling of dead cells with a DNA damage phenotype of (PARP-ve/H2AX+ve) and a reduction of parthanatos (PARP+ve/H2AX-ve, Figure. 4P). Dual blockade of shikonin’ s action with zVAD and necrostatin-1 resulted in a similar profile to that of untreated cells thus the mode of action of shikonin appears to be abrogated in terms of the DN phenotype (Figure. 4Q, R).
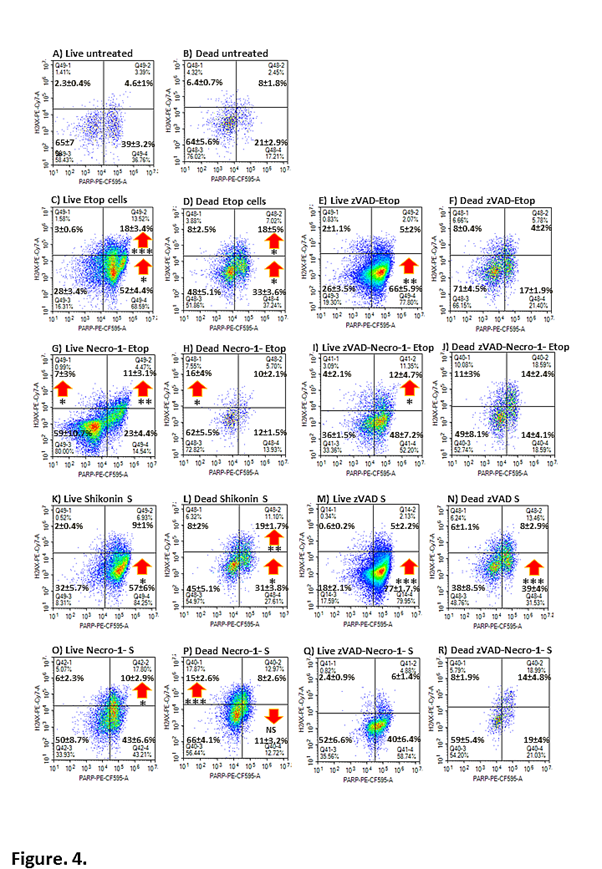
Figure 4: Double negative cells were further analysed for parthanatos and DNA damage. Jurkat cells were untreated or incubated with or without zVAD (20 μM) and/ or necrostatin-1 (60 μM) (Necro-1) with etoposide (Etop, 1 μM) or shikonin (S, 0.5 μM) for 24 h. Cells were labelled with the cell viability probe, Zombie NIR, fixed and permeabilised and then labelled with RIP3-PE, caspase-3-BV-650, PARP-PE-CFC595 and H2AX-PE-Cy7. Live/early apoptotic and dead cells undergoing parthanatos (lower right quadrant) or PARP+ve/H2AX-ve, DNA damage (upper left quadrant) or PARP-ve/H2AX+ve and parthanatos with DNA damage (upper right quadrant) by PARP+ve/H2AX+ve. Student T-test for comparisons of treated and untreated cells P<0.05 *, <0.01 **, <0.001 *** NS –not significant, n=3.
Conclusion
The use of a fixable live-dead cell probe with an active-caspase-3 antibody is more informative than the classic Annexin V assay commonly used in flow cytometry as it phenotypes dead cells into late apoptotic and necrotic9,17. The up-regulation of RIP3 within the live cell population is definitive for necroptosis but also allows the phenotyping of classic and RIP1-dependent apoptosis in live and dead cells13,16. The distribution of these populations in live and dead cells is significantly different indicating that Western Blot results from cell lysates will be potentially grossly affected by the relative distribution of live and dead cells in any such preparations. The addition of PARP and H2AX allows further dissection of RCD processes to a finer degree not shown by any other technical approach.
Future Directions
It has also been possible to further extend the assay by the addition of antibodies to detect autophagy (LC3B), ER stress (PERK) and pyroptosis (active Caspase-1 up-regulation). Interestingly autophagic cells were found to differ mainly in the level of DNA Damage which was reduced in live cells and increased in dead cells compared to untreated cells (data not shown). It is tempting to postulate that the autophagic process maintains cell health at the DNA level. Thus it is possible to simultaneously detect almost every form of RCD by this flow cytometric approach.
References
- Galluzzi L, Aaronson SA, Abrams J, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009; 16: 1093-107.
- Kepp O, Galluzzi L, Lipinski M, et al. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011; 10: 221-37.
- Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015; 22: 58-73.
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death and Differentiation. 2012; 19: 107-120.
- Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nature Reviews Cancer. 2015; 16: 20-33.
- Conrad M, Angeli JP, Vandenabeele P, et al. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016; 15: 348-66.
- Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015; 7.
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005; 73: 1907-16.
- Lee HL, Pike R, Chong MHA, et al. Simultaneous flow cytometric immunophenotyping of necroptosis, apoptosis and RIP1-dependent apoptosis. Methods. 2018; 134-135: 56-66.
- Jayaraman S. Intracellular determination of activated caspases (IDAC) by flow cytometry using a pancaspase inhibitor labeled with FITC. Cytometry A. 2003; 56: 104-12.
- Telford WG, Komoriya A, et al. Multiparametric Analysis of Apoptosis by Flow and Image Cytometry. Methods Mol Biology. 2011; 141-159.
- Pozarowski P, Huang X, Halicka DH, et al. Interactions of fluorochrome-labeled caspase inhibitors with apoptotic cells: a caution in data interpretation. Cytometry A. 2003; 55: 50-60.
- Fu Z, Deng B, Liao Y, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013; 13: 580-590.
- Wiench B, Eichhorn T, Paulsen M, et al. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid Based Complement Alternat Med. 2012; 2012: 726025.
- Sosna J, Voigt S, Mathieu S, et al. TNF?induced necroptosis and PARP?1?mediated necrosis represent distinct routes to programmed necrotic cell death. Cellular and Molecular Life Sciences. 2014; 71: 331-348.
- Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009; 137: 1112-23.
- Vermes I, Haanen C, Steffens-Nakken H, et al. A novel assay for apoptosis flow cytometric detection of phosphatidylserine early apoptotic cells using fluorescein labelled expression on Annexin V Journal of Immunological methods. 1995; 184: 39-51.