
Breaking up with ATM
Marek Adamowicz1*
1Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, Falmer, Brighton BN1 9RH, UK.
Abstract
ATM kinase is a master regulator of the DNA damage response (DDR). A recently published report from the d’Adda di Fagagna laboratory1 sheds a light onto our understanding of ATM activation. In this short-commentary we will expand on this and other work to perceive better some of the aspects of ATM regulation.
ATM and DNA damage response activation
ATM belongs to PI3K-like kinase family that consists of ATR, DNA-PKcs, mTOR, TRRAP and SMG1 kinases, which share similar structures and ability to phosphorylate their substrates on Serine and Threonine residues2. ATM was first discovered and cloned over 20 years ago in the laboratory of Yossi Shiloh3. ATM is recruited to DNA double strand breaks (DSBs) by MRN complex, composed of MRE11, RAD50 and NBS1 proteins, which senses DNA damage and quickly localizes to the DSBs. This process induces ATM activation and triggers a downstream cascade that leads to the engagement of DDR factors like: MDC1 and 53BP1, around DNA lesions (Figure 1).
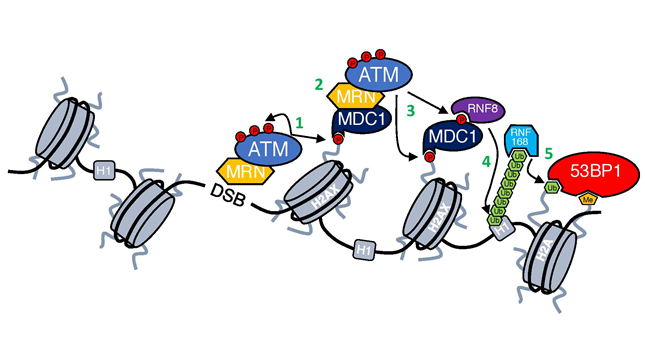
Figure 1: DNA damage response cascade.
First step in the DDR activation is recruitment of ATM kinase by MRN complex to DSBs through its interaction with NBS14,5. At the DSB ATM is activated (for details see Figure 2) and phosphorylates both itself and other substrates (1). One of the most important substrates of ATM is H2AX6, which provides a scaffold for the further accumulation of DDR factors7. γH2AX is recognized and bound by MDC1 that then enables further accumulation of ATM-MRN complex and spreading of γH2AX around DSBs (2)8,9. ATM-mediated phosphorylation of MDC1 allows also for the recruitment of RNF8 ubiquitin ligase (3), which ubiquitinates histone H1 and enables the recruitment of another ubiquitin ligase RNF168 (4)10,11, which in turn ubiquitinates histone H2A12. Ubiquitination of H2A together with the deposition of methyl group done by MMSET13 results in the recruitment of 53BP1 (5), which coordinates with other factors DNA repair pathway choice14.
Upon recruitment to DSBs, ATM dimer undergoes a complicated process of activation. First, it is acetylated by lysine acetyltransferase 5 (KAT5/TIP60) at Lysine 301615, which is followed by autophosphorylation, in-trans on Serine 1981, leading to its monomerization and conferring access of ATM substrates to the kinase domain16,17(Figure 2A).
In recent work, Adamowicz et al., show that ATM localizes to the DSBs in a protein complex together with FOXO3a and KAT5/Tip60 (from now on referred to as AAC (ATM Activation Complex)). Formation of this complex is dependent on FOXO3a bridging an interaction between ATM and KAT5 (Figure 2A and B). Formation of AAC is inhibited by a well-known transcription factor, NOTCH1, which is a negative regulator of ATM activation through its ATM binding ability24 (Figure 2B). Interestingly this leads to MRN-mediated recruitment of ATM to DSBs without the latter being activated at the site of the damage, as observed by the lack of autophosphorylation (pSer1891ATM)1. Strikingly, although all downstream DDR events are attenuated due to NOTCH1-mediated ATM inactivation (including ATM-mediated phosphorylations like: p53, CHK2, SMC1), phosphorylation of the ATM main substrate, histone H2AX (Ser139H2AX, from now on referred as γH2AX) is not affected1,24. Below I will try to focus on those two aspects of ATM activation (ATM autophosphorylation and phosphorylation of H2AX), putting them in the context of already published reports. At the end, I will speculate about the role of NOTCH-FOXO3a competition in cells and possible future areas of ATM research.
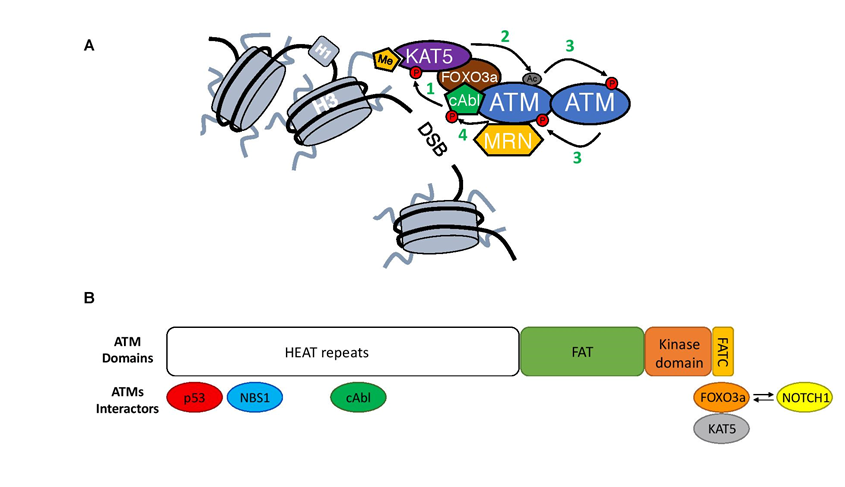
Figure 2: Close-up on ATM activation.
A) Upon MRN-mediated recruitment to DSBs ATM undergoes an activation process. First, cAbl kinase phosphorylates KAT5 on Tyrosine 44 (1)18, which together with KAT5 interaction with H3K9m3 leads to KAT5 activation19. This way stimulated KAT5 will mediate acetylation of ATM in its PRD domain (Lysine 3016)(2)15,20. As a consequence of acetylation, ATM undergoes structural changes in its dimer form that allows for the phosphorylation in trans each of ATM monomers (3)16,20. ATM autophosphorylation leads to its monomerization and full activation16. At the end, activated ATM will generate a positive feedback loop and phosphorylate cAbl kinase leading to its increased activation (4)21.
B) Structure of the ATM kinase (1-3056aa; Domains: FAT 1966–2566aa; kinase domain 2614–2960aa and FATC 3025–3056aa) together with its most important interactors and their binding sites1,22-25. Competition between FOXO3a and NOTCH1 for the binding to the FATC domain is depicted with arrows.
ATM phosphorylation – is it still a thing?
ATM phosphorylation on Serine 1981 is commonly used as a marker for activated ATM. ATM phosphorylation was first observed almost 20 years ago26,27 and since then many of ATM’s autophosphorylation sites have been identified28,29. It has been established that ATM autophosphorylation on Serine 1981 is necessary for ATM monomerization and kinase activation16,20. Additionally it has been shown that ATM mutants carrying the S1981A substitution do not rescue radio-sensitivity of the A-T cells in transfection experiments16. In agreement with those reports we have shown that NOTCH1-mediated inhibition of ATM autophosporylation resulted in the attenuation of phosphorylation of ATM downstream substrates and checkpoint impairment1,24. On the other hand it has been reported that mouse Atm S1987A mutants (mouse equivalent of human S1981A) are proficient in DDR and ATM kinase activity30. Moreover, it seems that other Atm autophosphorylation sites (S367 and S1893) are dispensable for Atm activation31. Interestingly in the in vitro kinase assay ATM S1981A mutant can phosphorylate CHK2 and p53 in response to linear dsDNA to the same extent as the wild-type ATM32. Although we observed NOTCH1 inhibiting ATM-mediated p53 phosphorylation in vitro24, in the light of the new results demonstrating disruption of KAT5 and FOXO3a from the AAC1 it would be interesting to see if addition of: KAT5, FOXO3a, H3K9m3 and cAbl would give similar results in an in vitro ATM kinase assay in the presence of NOTCH1 and mutated ATM (S1981A).
It has been suggested that ATM autophosphorylation is necessary for ATM retention at the DSBs33 because the ATM S1981A mutant, although recruited normally to DSBs, was not stabilized properly at the site of the damage33. In contrast we have reported that although NOTCH1 inhibited ATM autophosphorylation it did not affect ATM’s retention at the DSBs1. To understand these apparently contradictory observations one should look not just at the ATM autophosphorylation. We have previously reported that NOTCH1 inhibits kinase activity of ATM24, therefore it would be better to compare our results with the reports describing recruitment of the ATM kinase dead mutant (ATM KD). Indeed it has been shown that ATM KD mutant (that cannot undergo autophosphorylation) is recruited to the DSBs without any impairment of retention34,35. This is similar to our observation of the NOTCH1-mediated impact on ATM recruitment and autophosphorylation. Additionally, it was reported that cells carrying mutations in the KU70 and MRE11 nuclease actively recruit ATM to the DSBs (without retention impairment); although ATM does not undergo autophosphorylation in those conditions36. Overall, our results1,24, together with above mentioned reports, show our still incomplete understanding of ATM autophosporylation and its role in the ATM activity and DDR, that needs further elucidation.
Meeting γH2AX at the FATC end
We have repeatedly observed that NOTCH1 inhibits ATM activation by blocking its autophosphorylation (Ser1981) and phosphorylation of downstream substrates such as KAP1, SMC1, p53, DNA-PKcs or CHK21,24(Adamowicz et al – in press). Additionally, we have observed in our initial study using Xenopus laevis egg extract that NOTCH1 blocked a substantial amount of ATM-mediated phosphorylation24. However, of all observed NOTCH1-mediated ATM phosphorylation defects, phosphorylation of ATM’s main substrate, H2AX, remained unaffected.
It has been suggested that there is redundancy between PI3K-like kinases in terms of H2AX phosphorylation. DNA-PKcs or ATR kinases have been shown to phosphorylate H2AX37,38. Additionally, it has been reported that H2AX is phosphorylated to the same extent in the ATM WT and KO cells37,39. Indeed, experiments carried out in our laboratory showed that NOTCH1 neither blocks ATR nor DNA-PKcs kinase activity (Adamowicz et al – in press). However unexpectedly, when we performed analysis of the γH2AX foci formation in NOTCH1-expressing cells in the presence of either DNA-PKcs or ATR inhibitors we did not observe any difference in the H2AX phosphorylation (data unpublished – data available upon request).
These results suggest that H2AX can be phosphorylated by protein kinases other than ATM, ATR and DNA-PKcs. Indeed, it has been reported that JNK and p38 can phosphorylate H2AX in response of UV light irradiation or starvation respectively40,41. Additionally, it has been reported that VRK1 kinase can phosphorylate H2AX in response to IR in parallel to ATM kinase42. Moreover, VRK1 was shown to be necessary for the accumulation of DDR factors around DSBs, which implies more complex role of VRK1 in the DDR that needs further elucidation43.
Because NOTCH1, unlike small molecule ATM inhibitors, cannot directly inhibit ATM kinase activity we can speculate that NOTCH1 binding to ATM could strongly impair ATM substrate recognition, resulting in an inhibition of phosphorylation of some substrates such as p53 or CHK2, but not H2AX. It has been already reported that NOTCH1 can bind and hence modulate the substrate recognition of LSD1 demethylase44. Therefore, it can be possible that by binding to the FATC domain of ATM, NOTCH1 would strongly impair substrate recognition of ATM. Interestingly, it has been shown in yeast that deletion of last 10 amino acids (aa) of the FATC domain can impair Tel1 phosphorylation of Rad53, which was connected with the loss in its ability to interact with MRX complex45. Moreover, MRN complex was shown to help ATM in the substrate recognition by stimulating ATM binding to its substrates like p53 or CHK246. On the other hand it has been published that in human cells deletion of last 10aa of the FATC domain of ATM does not lead to the impairment of ATM MRN-mediated response, but rather its ability to activate upon oxidative stress47. We have shown that although NOTCH1 binds to the FATC domain it does not affect interaction between ATM and MRN complex1. This suggests that if by binding to the ATM FATC domain NOTCH1 is perturbing ATM substrate recognition this effect is rather mediated by inhibition of KAT5-mediated acetylation. Impairment of ATM acetylation will then block structural changes in ATM that would lead to its monomerization and activation, inhibiting this way release of the ATM kinase domain otherwise hindered inside of its dimer structure17.
Taking ATM down a NOTCH
NOTCH1 was very early connected to the tumorogenesis and marked as an oncogene due to its ability to induce tumour growth48. Activating mutations in NOTCH1 are present in many T-cell acute lymphoblastic leukemias (T-ALL)49 or breast cancers50. Indeed, we have found that NOTCH1 expression was negatively correlated with the ATM activation in the human breast cancer patients24. At the same time, other group reported that ectopic expression of NOTCH1 in cancer cells lead to their increased resistance to DNA damage in vivo51. Those data show that expression of high levels of NOTCH1 (due to activating mutations or ectopic expression) stimulate radioresistance and survival in cancer cells, resulting probably from the inhibition of p53-mediated apoptosis. Additionally, increased levels of NOTCH1 induce faster proliferation52 leading to replication stress. Although we showed that NOTCH1 inhibits ATM activation, this is not true for ATR kinase (Adamowicz et al – in press) resulting in the protection of NOTCH1-driven cancers from replication stress.
Neural stem cells (NSC) are known to express moderate levels of activated NOTCH1, which is necessary for their proliferation53. Interestingly it has been reported that induction of DDR in NSC leads to their spontaneous differentiation to astrocytes, which is dependent on ATM activation54. It is therefore possible to speculate that NOTCH1-mediated downregulation of ATM activation could tip the balance allowing for DNA damage repair without inducing differentiation. It is important to remember that physiological levels of NOTCH1 are low as compared to those observed in T-ALL cells or those achieved by ectopic expression, therefore observed effects of NOTCH1 activation might be very mild. Additionally, observed results might be an outcome of many different factors impacting at the same time on DDR. Indeed, it has been shown that SALL4 transcription factor expressed in stem cells favours ATM activation by its binding to MRN complex55.
In summary, I would like to propose that the physiological role of NOTCH1 is not to inhibit fully ATM activation, but rather to induce its mild impairment, to modulate a balance between the amount of DNA damage and DDR signalling. This would result in the suppression of DNA damage induced apoptosis or differentiation, giving time for necessary repair.
There is plenty more ATM in the sea
Formation of AAC is necessary for ATM activation at DSBs and DDR1,24. Apart from AAC, ATM relies also on MRN complex, which allows proper AAC localization and substrate recognition. Interestingly, in the nucleus, ATM has been described to exist in two different complexes. In has been shown that there is competition between MRN complex and ATMIN for binding to ATM (Figure 3)56,57. Studies have found that while MRN complex guides ATM in the response to DSBs, ATMIN is necessary during oxidative and hypotonic stresses58,59. It is possible that like MRN, ATMIN by binding to ATM regulates it substrate recognition and therefore its kinase activity in response to different stimuli. It would be interesting to see if the structure of AAC is preserved while complexed with ATMIN, and if so how it is involved in the ATM activation (Figure 3).
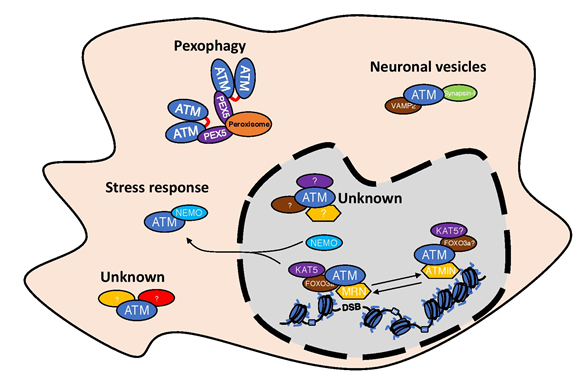
Figure 3: Existence of different ATM complexes in the cells.
Schematic representation of the existence of different ATM complexes both in the nucleus as well as in the cytosol. For details please look into the text.
We tend to think about ATM through its role in DDR regulation, although ATM has been described to be involved in many more cellular processes like: stress response60, neuronal signal transmission61 or pexophagy62,63(Figure 3), which in every case requires its presence outside the nucleus. This implies that ATM is not always in complex with KAT5 or MRN complex. The presence of ATM in peroxisomes is a result of its interaction with PEX5, which is responsible for ATM peroxisome localization. In peroxisomes, ATM is activated by reactive oxygen species and formation of an active dimer, allowing ATM to control peroxisome phagocytosis63. The involvement of ATM in the stress response is connected to its interaction with NEMO and with shuttling between nucleus and cytosol60. Additionally, ATM has been described to localize in the cytosol of neuronal cells64 and has been implicated in the neuronal signal transmission by its interaction with VAMP2 and Synapsin-I61. It would be interesting to see if NOTCH1, which has very strong affinity to the FATC domain of ATM1, could be used as a tool for identifying new regulatory components of ATM complexes in the cytosol.
It is thought-provoking to picture ATM in different complexes that differentially regulate its activity and substrate recognition. The identification of different active ATM complexes opens new and exciting areas of research and raises even more fascinating questions. For example, how is ATM activation in those complexes stimulated, and how is ATM substrate recognition and kinase activity regulated? Hopefully, in the near future we will know the answers.
Acknowledgements
I thank Sheetal Sharma, Jelena Vermezovic and Keith Caldecott for their critical reading of the manuscript. I apologize to all the authors whose work I could not include due to the space constrains of this short-commentary/review.
Conflict of Interest
I declare no conflict of interest.
References
- Adamowicz M, Vermezovic J, d'Adda di Fagagna F. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016; 16: 2068-2076.
- Paull TT. Mechanisms of ATM Activation. Annual review of biochemistry. 2015; 84: 711-738.
- Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995; 268: 1749-1753.
- Falck,J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005; 434: 605-611.
- Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003; 17: 1957-1962.
- Burma S, Chen BP, Murphy M, et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. The Journal of biological chemistry. 2001; 276: 42462-42467.
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biology. 2003; 5: 675-679.
- Stucki M, Clapperton JA, Mohammad D, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005; 123: 1213-1226.
- Chapman JR, Jackson, SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO reports. 2008; 9: 795-801.
- Thorslund T, Ripplinger A, Hoffmann S, et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015; 527: 389–393.
- Mailand N, Bekker-Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007; 131: 887-900.
- Doil C, Mailand N, Bekker-Jensen S, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009; 136: 435-446.
- Pei H, Zhang L, Luo K, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011; 470: 124-128.
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nature reviews. Molecular cell biology. 2014; 15: 7-18.
- Sun Y, Jiang X, Chen S, et al. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005; 102: 13182-13187.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003; 421: 499-506.
- Sawicka M, Wanrooij PH, Darbari VC, et al. The dimeric architecture of checkpoint kinases Mec1ATR and Tel1ATM reveal a common structural organisation. The Journal of biological chemistry. 2016; 291: 13436-13447.
- Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013; 498: 70-74.
- Sun Y, Jiang X, Xu Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol. 2009; 11: 1376-1382.
- Sun Y, Xu Y, Roy K, et al. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007; 27: 8502-8509.
- Baskaran R, Wood LD, Whitaker LL, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997; 387: 516-519.
- Pessina F, Lowndes NF. The RSF1 Histone-Remodelling Factor Facilitates DNA Double-Strand Break Repair by Recruiting Centromeric and Fanconi Anaemia Proteins. PLoS Biology. 2014; 12.
- Khanna K, Keating KE, Kozlov S, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nature Genetics. 1998; 20: 398-400.
- Vermezovic J, Adamowicz M, Santarpia L, et al. Notch is a direct negative regulator of the DNA-damage response. Nat Struct Mol Biol. 2015; 22: 417-424.
- Jiang X, Sun Y, Chen S, et al. The FATC Domains of PIKK Proteins Are Functionally Equivalent and Participate in the Tip60-dependent Activation of DNA-PKcs and ATM. Journal of Biological Chemistry. 2006; 281: 15741-15746.
- Kim ST, Lim DS, Canman CE, et al. Substrate specificities and identification of putative substrates of ATM kinase family members. The Journal of biological chemistry. 1999; 274: 37538-37543.
- Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science (New York, N.Y.). 1998; 281: 1677-1679.
- Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science (New York, N.Y.). 2007; 316: 1160-1166.
- Kozlov SV, Graham ME, Peng C, et al. Involvement of novel autophosphorylation sites in ATM activation. The EMBO journal. 2006; 25: 3504-3514.
- Pellegrini M, Celeste A, Difilippantonio S, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006; 443: 222-225.
- Daniel JA, Pellegrini M, Lee JH, et al. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. The Journal of cell biology. 2008; 183: 777-783.
- Lee JHH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005; (New York, N.Y.) 308: 551-554.
- So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009; 187: 977-990.
- Daniel JA, Pellegrini M, Lee BS, et al. Loss of ATM kinase activity leads to embryonic lethality in mice. The Journal of cell biology. 2012; 198: 295-304.
- Yamamoto K, Wang Y, Jiang W, et al. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol. 2012; 198: 305-313.
- Hartlerode AJ, Morgan MJ, Wu Y, et al. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat Struct Mol Biol. 2015; 22: 736-743.
- Stiff T, O'Driscoll M, Rief N, et al. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer research. 2004; 64: 2390-2396.
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. The Journal of biological chemistry. 2001; 276: 47759-47762.
- Riballo E, Kühne M, Rief N, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Molecular cell. 2004; 16: 715-724.
- Lu C, Shi Y, Wang Z, et al. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS letters. 2008; 582: 2703-2708.
- Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Molecular cell. 2006; 23: 121-132.
- Salzano M, Sanz-García M, Monsalve DM, et al. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics. 2015; 10: 373-383.
- Sanz-García M, Monsalve DM, Sevilla A. et al. Vaccinia-related kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. The Journal of biological chemistry. 2012; 287: 23757-23768.
- Yatim A, Benne C, Sobhian B, et al. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol Cell. 2012; 48: 445-458.
- Ogi H, Goto GH, Ghosh A, et al. Requirement of the FATC domain of protein kinase Tel1 for localization to DNA ends and target protein recognition. Mol Biol Cell. 2015; 26: 3480-3488.
- Lee JHH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science (New York, N.Y.). 2004; 304: 93-96.
- Guo Z, Kozlov S, Lavin MF, et al. ATM activation by oxidative stress. Science (New York, N.Y.). 2010; 330: 517-521.
- Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. The Journal of Experimental Medicine. 1996; 183: 2283-2291.
- Weng AP, Ferrando AA, Lee W, et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science. 2004; 306: 269-271.
- Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. The Journal of Cell Biology. 2004; 167: 215-221.
- Theys J, Yahyanejad S, Habets R et al. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiotherapy and Oncology. 2013; 108: 440-445.
- Mazzone M Selfors LM, Albeck J, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107: 5012-5017.
- Hitoshi S, Alexson T, Tropepe V, et al. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes & Development. 2002; 16: 846-858.
- Schneider L, Pellegatta S, Favaro R, et al. DNA Damage in Mammalian Neural Stem Cells Leads to Astrocytic Differentiation Mediated by BMP2 Signaling through JAK-STAT. Stem Cell Reports. 2013; 1: 123-138.
- Xiong J, Todorova D, Su NY, et al. Stemness factor Sall4 is required for DNA damage response in embryonic stem cells. The Journal of Cell Biology. 2015; 208: 513-520.
- Zhang T, Cronshaw J, Kanu N, et al. UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation-induced ATM signaling and function. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111: 12091-12096.
- Zhang T, Penicud K, Bruhn C, et al. Competition between NBS1 and ATMIN controls ATM signaling pathway choice. Cell reports. 2012; 2: 1498-1504.
- Kanu N, Behrens A. ATMIN defines an NBS1-independent pathway of ATM signalling. The EMBO journal. 2007; 26: 2933-2941.
- Kanu N, Penicud K, Hristova M, et al. The ATM cofactor ATMIN protects against oxidative stress and accumulation of DNA damage in the aging brain. The Journal of biological chemistry. 2010; 285: 38534-38542.
- Wu ZHH, Shi Y, Tibbetts RS, et al. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science (New York, N.Y.). 2006; 311: 1141-1146.
- Li J, Han YR, Plummer MR, et al. Cytoplasmic ATM in neurons modulates synaptic function. Current biology : CB. 2009; 19: 2091-2096.
- Watters D, Kedar P, Spring K, et al. Localization of a portion of extranuclear ATM to peroxisomes. The Journal of biological chemistry. 1999; 274: 34277-34282.
- Zhang J, Tripathi DN, Jing J, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nature cell biology. 2015; 17: 1259-1269.
- Barlow C, Ribaut-Barassin C, Zwingman TA, et al. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97: 871-876.