
Commentary: Small Molecule Inhibition of PD-1 Transcription is an Effective Alternative to Antibody Blockade in Cancer Therapy
Alison Taylor1*, Christopher E. Rudd2,3
1Leeds Institute of Medical Research, University of Leeds, School of Medicine, Wellcome Trust Brenner Building, St James’s University Hospital, LEEDS LS9 7TF, UK
2Division of Immunology-Oncology Research Center, Maisonneuve-Rosemont Hospital, Montreal, Quebec H1T 2M4, Canada
3Département de Medicine, Université de Montréal, Montreal, Quebec H3C 3J7, Canada
The past few years has witnessed exciting progress in the application of “immune check-point inhibitors” (ICI) in the treatment of various human cancers1-3. This involves the use of antibody blockade with monoclonal antibodies (mAbs) that block receptor binding to their natural ligands. Programmed cell death-1 (PD-1) recognises PD ligand (PDL)-1 and PDL-2 on presenting cells and this sends signals that inhibit T-cell activation and effector cytotoxic responses. Through these mechanisms, PD-1 inhibits the immune system and can prevent autoimmune diseases 4. Tumor cells expressing PDL-1/PD-L2 can use this mechanism to evade immune surveillance, allowing disease progression. A therapeutic approach involves administration of mAbs that block the engagement of checkpoint molecules with their ligand. In the case of anti-PD-1, these mAbs block the binding of PD-1 on the T-cell with PDL-1/PDL-2 on the tumor cell, preventing recognition and allowing activation of the T-cell to provide an immune response against the tumor cell. Blockade also reverses T-cell exhaustion and restores T-cell functionality 5, 6. Furthermore, PD-1 expression on tumor-infiltrating CD8+ T-cells correlates with impaired function, while PD-L1 expression on tumors facilitates escape4.
One of the first established immunotherapeutic approaches involved the use of Ipilimumab against CTLA-47, 8. It was the prototypical immunomodulatory antibody first approved by the FDA in 2011 for advanced melanoma based on its survival benefit. This was followed by the highly successful blockade of PD-1 (i.e. Nivolumab and Pembrolizumab), or its ligand (PD-L1) (i.e. Atezolizumab), either alone7, or in combination with anti-CTLA-48. In certain cases, the use of PD-1 mAbs superseded CTLA-4 mAbs, due to their increased response rates9, 10 and the combination of both therapies gave rise to even superior response rates10, 11. However, this success correlated with increased toxic side effects. A substantial proportion of patients receiving ICI develop immune-related adverse events (irAEs) including colitis, endocrinopathies, hepatitis, pneumonitis, cardiotoxicity, nephritis, skin eruptions and vitiligo12-20. These events have been reported at 20-28%, 17-21% and 45-59% for the use of anti-CTLA-4, anti-PD1 or combination therapy, respectively9-11. These drugs are currently being used in the treatment of various cancers including Melanoma, renal cell carcinoma, colorectal cancer and Hodgkin lymphoma21-24 as well as the viral infection HCV25. Immune-modulating agents, such as corticosteroid, infliximab, and mycophenolic acid are being used to manage irAEs26 where possible but in some cases, treatment is discontinued.
Although some success has been seen, the majority of patients are still not cured, some develop resistance and those with immune-resistant cancers such as colon and ovarian are poorly responsive. This poor prognosis highlights a need to improve current or identify alternative clinical interventions.
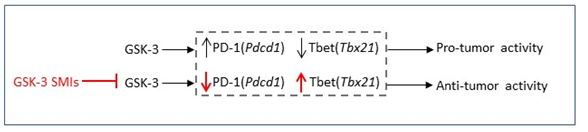
As PD-1 plays a prominent role in immunotherapy, one approach for enhanced anti-tumor immunity would be to inhibit pathways that control the expression of inhibitory co-receptors such as PD-1. We are the first to show that the serine/threonine kinase glycogen synthase kinase 3 (GSK3) is a central regulator of PD-1 expression in CD8+ T cells.
There are two isoforms of GSK-3, GSK-3α and GSK-3β, which are encoded by separate genes, with highly homologous kinase domains (98% identity) but divergent N- and C-terminal regions27, 28. Both forms have been implicated in processes ranging from glycogen metabolism to gene transcription, apoptosis and microtubule stability. The notable aspect of GSK-3 is that it is constitutively active in resting T-cells and is inhibited by receptor induced activation signals29.
In this regard, we have shown that small molecule inhibitors (SMIs) of GSK-3 are effective in promoting viral clearance30 and our current work31 shows that GSK-3 SMI inhibition of Pdcd1 (PD-1) transcription with a small molecule inhibitor (i.e. SB415286) is as effective as anti-PD-1 and PDL-1 blocking antibodies in the control of B16 and EL-4 tumor growth. Similar effects were observed using other inhibitors including SB216763 and CHIR99021 as well as the peptide inhibitor L803-mts. The exception was the inhibitor TWS119 which has been reported to retain cells in a less mature state32,33, by promoting the expression of TCF-1, blocking CD8+ T-cell differentiation, and inhibiting IFN-γ production32,34. Whereas other SMIs including SB415286 have been seen to promote differentiation and IFNγ production30,35-36. This difference between SMIs in their action on T-cell function underlines the need for defining the pathways of GSK-3 in T-cell signaling.
Our current work demonstrated that SB415286 significantly reduced B16 pulmonary metastasis. This anti-tumor effect of SB415286 was comparable to that using anti-PD-1 blocking antibody and combination of the two had no additional effect indicating an overlap in the two pathways. Further to this GSK-3 deficient T-cells from conditional knockout mice significantly reduced tumor progression confirming a direct role for GSK-3 in modulating anti-tumor activity in CD8+ T-cells. Our findings showed that GSK-3 inhibition operated primarily via a reduction in PD-1 expression on CD8+ T-cells. Inactivation of GSK-3 either through SMIs or by using GSK-3α/β siRNA led to a reduction in PD-1 expression and in both cases reduced B16 pulmonary metastasis to a similar extent as seen in Pdcd1-/- mice. In each model, GSK-3 SMIs inhibited Pdcd1 transcription and PD-1 expression on tumor infiltrating T-cells (TILs), while increasing Tbx21 (T-bet) transcription30 and the presence of CD8+ TILs expressing CD107a (LAMP1), granzyme B (GZMB) and IFNγ 131. Other transcription factors such as Eomes (Eomesodermin) or the high mobility group (HMG) box Transcription factor 7 (Tcf7) were not affected.
Mechanistically, GSK-3 inactivation in T-cells with down-regulated T-bet had no effect on PD-1 expression indicating that GSK-3 operates upstream and is dependent on T-bet which in turn inhibits PD-1 expression. Further, down-regulation of T-bet increased PD-1 transcription indicating that T-bet suppresses the transcription of PD-1, in accord with results from the Wherry group37. High levels of T-bet expression could sustain exhausted CD8+ T-cells and repressed the expression of inhibitory receptors during chronic viral infection. Persistent antigenic stimulation caused downregulation of T-bet, which resulted in more severe exhaustion of CD8+ T-cells37.
Overall, in our model, active GSK-3 present in resting T-cells acts to supress Tbx21 transcription. Upon T-cell activation GSK-3 becomes partially inactivated leading to partial T-bet expression and PD-1 suppression. The use of GSK-3 SMIs can fully repress GSK-3 leading to increased T-bet expression and complete inhibition of PD-1 expression.
The development of small molecules that modulate co-receptors or their signaling pathways to enhance anti-tumor activity would be a major advance in therapy. There are potential advantages and disadvantages to the use of GSK-3 inhibitors versus anti-PD-1 antibody therapies. Small molecules have the advantage of lower cost, dosing and potential oral administration. Further, anti-PD-1 is associated with a high cost as well as adverse effects such as fatigue, rash and possible autoimmune complications such as colitis. Although we cannot exclude these effects with GSK-3 SMIs, to date, we have seen no evidence of autoimmunity in the GSK-3α/β-/- mice over 2 years. The disadvantage of GSK-3 inactivation is the potential of an effect on the function of other host cells or the tumor itself. However, lithium chloride, another GSK-3 inhibitor, has been used for decades for the treatment of bipolar disease. Importantly, we showed that GSK-3 inhibition could affect PD-1 expression on both murine and human T-cells. The dose (200ug per 20g mouse) used is roughly comparable to the dose of another inhibitor Tideglusib which was used in a phase 2 oral study (800mg in a 80kg patient) to treat progressive supranuclear palsy38. Further, we showed that a single dose of SB415286 down-regulated PD-1 for 10-14 days. Although, we failed to see any effect of SB415286 directly on the growth of B16 melanoma cells in the absence of an immune response, GSK-3 inhibition has been reported to directly inhibit the growth of multiple myeloma, neuroblastoma, hepatoma and prostate tumors38-43. It is therefore possible that GSK-3 SMIs might have an added advantage by directly inhibiting the growth of some tumors in addition to enhancing the immune response. Despite these possibilities, the major effect of GSK-3 SMIs in our studies involved the amplification of the immune system as shown by the effects on ex vivo T-cells and adoptive transfer experiments as well as by the elimination of tumors in mice where T-cells have conditionally deleted GSK-3α/β. Certain tumors can impair proximal TCR signaling events as a form of immune avoidance44,45. The inhibition of GSK-3 could potentially circumvent this impairment given that GSK-3 operates down-stream of proximal signal mediators such as p56lck. Overall, our findings identify a potential alternate approach using small molecule inhibition of PD-1 expression in cancer immunotherapy. Further work is needed to uncover the full range of down-stream effects that may be regulated by GSK-3 regulation in anti-tumor immunity.
References
- Page DB. Immune modulation in cancer with antibodies. Annu Rev Med. 2014; 65: 185-202.
- Pentcheva-Hoang T, Simpson TR, Montalvo-Ortiz W, et al. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res. 2014; 2(10): 970-80.
- Sharma P, Wagner K, Wolchok JD, et al. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011; 11(11): 805-12.
- Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000; 192(7): 1027-34.
- Freeman GJ, Wherry EJ, Ahmed R, et al. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. The Journal of experimental medicine. 2006; 203: 2223-2227.
- Ha SJ, Mueller SN, Wherry EJ, et al. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. J Exp Med. 2008; 205: 543-555.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366(26): 2443-54.
- Wolchok JD. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369(2): 122-33.
- Schachter J, Ribas A, Long GV, et al., Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. 2017; 390(10105): 1853-1862.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2017; 377(14): 1345-1356.
- Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017; 18(9): 1202-1210.
- Arai T, Harada K, Usui Y, et al., Case of acute anterior uveitis and Vogt-Koyanagi-Harada syndrome-like eruptions induced by nivolumab in a melanoma patient. J Dermatol. 2017; 44(8): 975-976.
- Day D, Hansen AR. Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitors. BioDrugs. 2016; 30(6): 571-584.
- De Velasco G, Je Y, Bossé D, Awad MM,, et al., Comprehensive Meta-analysis of Key Immune-Related Adverse Events from CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res. 2017; 5(4): 312-318.
- Fujimura T, Kambayashi Y, Furudate S, et al. Isolated adrenocorticotropic hormone deficiency possibly caused by nivolumab in a metastatic melanoma patient. J Dermatol. 2017; 44(3): e13-e14.
- Heinzerling L, Ott PA, Hodi FS, et al., Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J Immunother Cancer. 2016; 4: 50.
- Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: A multi-institutional retrospective study. J Dermatol. 2017; 44(2): 117-122.
- Tanaka A, Ikinaga K, Kiyohara E, et al. Critical renal adverse event induced by nivolumab therapy in a stage IV melanoma patient. J Dermatol. 2017; 44(6): 727-728.
- Teramoto Y, Nakamura Y, Asami Y, et al., Case of type 1 diabetes associated with less-dose nivolumab therapy in a melanoma patient. J Dermatol. 2017; 44(5): 605-606.
- Wada N, Uchi H, Furue M. Case of remitting seronegative symmetrical synovitis with pitting edema (RS3PE) syndrome induced by nivolumab in a patient with advanced malignant melanoma. J Dermatol. 2017; 44(8): e196-e197.
- Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008; 14(10): 3044-51.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015; 372(26): 2521-32.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014; 32(10): 1020-30.
- Westin JR, Chu F, Zhang M, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014; 15(1): 69-77.
- Gardiner D, Lalezari J, Lawitz E, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013; 8(5): e63818.
- Weber JS, Hodi FS, Wolchok JD, et al. Safety Profile of Nivolumab Monotherapy: A Pooled Analysis of Patients With Advanced Melanoma. J Clin Oncol. 2017; 35(7): 785-792.
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001; 359: 1-16.
- Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. The EMBO journal. 1990; 9: 2431-2438.
- Woodgett JR. Judging a protein by more than its name: GSK-3. Science's STKE : signal transduction knowledge environment. 2001; 2001(100): re12.
- Taylor A, Harker JA, Chanthong K, et al. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of co-receptor PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity. 2016; 44(2): 274-86.
- Taylor A, Rothstein D, Rudd CE, et al., Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018; 78(3): 706-717.
- Gattinoni L, Zhong XS, Palmer DC, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009; 15: 808–813.
- Philip M, Fairchild L, Sun L, et al. Chromatin states define tumor-specific T cell dysfunction and reprogramming Nature. 2017; 545: 452.
- Tang YY, Sheng SY, Lu CG, et al. Effects of Glycogen Synthase Kinase-3β Inhibitor TWS119 on Proliferation and Cytokine Production of TILs From Human Lung Cancer. J Immunother. 2018; 41: 319–328.
- Schroeder JH, Bell LS, Janas ML, et al. Pharmacological Inhibition of Glycogen Synthase Kinase 3 Regulates T Cell Development In Vitro. PLos ONE. 2013; 8(3): e58501.
- Coghlan MP, Culbert AA, Cross DA, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000; 7(10): 793-803.
- Kao C, Oestreich KJ, Paley MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011; 12(7): 663-71.
- Tolosa E, Litvan I, Höglinger GU, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014; 29(4): 470-8.
- Zhu Q, Yang J, Han S, et al. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate. 2011; 71(8): 835-45.
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996; 93(16): 8455-9.
- Piazza F, Manni S, Tubi LQ, et al. Glycogen Synthase Kinase-3 regulates multiple myeloma cell growth and bortezomib-induced cell death. BMC Cancer. 2010; 10: 526.
- Dickey A, Schleicher S, Leahy K, et al. GSK-3beta inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J Neurooncol. 2011; 104(1): 145-53.
- Beurel E, Blivet-Van Eggelpoël MJ, et al. Glycogen synthase kinase-3 inhibitors augment TRAIL-induced apoptotic death in human hepatoma cells. Biochem Pharmacol. 2009; 77(1): 54-65.
- Finke JH, Zea AH, Stanley J, et al. Loss of T-cell receptor zeta chain and p56lck in T-cells infiltrating human renal cell carcinoma. Cancer Res. 1993; 53(23): 5613-6.
- Rayman P, Uzzo RG, Kolenko V, et al., Tumor-induced dysfunction in interleukin-2 production and interleukin-2 receptor signaling: a mechanism of immune escape. Cancer J Sci Am. 2000; 6 Suppl 1: S81-7.