
Detection of Antigen-Specific T Cell Lineages and Effector Functions Based on Secretory Signature
Greg A. Kirchenbaum, Jodi Hanson, Diana R. Roen, Paul V. Lehmann*
Cellular Technology Limited (CTL) Shaker Heights, OH, 44122-5350, USA
Abstract
T cells not only protect us from infectious diseases and cancer, but are also involved in transplant rejection, autoimmune diseases, and allergies. Each of these immunologic processes share a common link in which antigen-specific T cells undergo expansion, with some of the resulting progeny differentiating into memory cells. Memory T cells belong to several distinct lineages, and sub-lineages, that fundamentally differ in their effector functions and capacity to mediate a protective or pathological immune response. In this mini-review, we outline how such memory T cell subpopulations can readily be identified on the basis of their secretory signature using a multi-color ImmunoSpot® assay.
The human immune system is composed of both an innate and adaptive arm (Figure 1). Innate cells, such as macrophages and neutrophils, serve as the first line of defense against foreign invaders by recognizing common features of pathogens using a limited number of conserved pattern recognition receptors1,2. While the innate arm of the immune system conveys protection against most environmental microorganisms, it is unable to recognize unique features of pathogens, possesses limited effector functions, and is unable to generate immunological memory1.
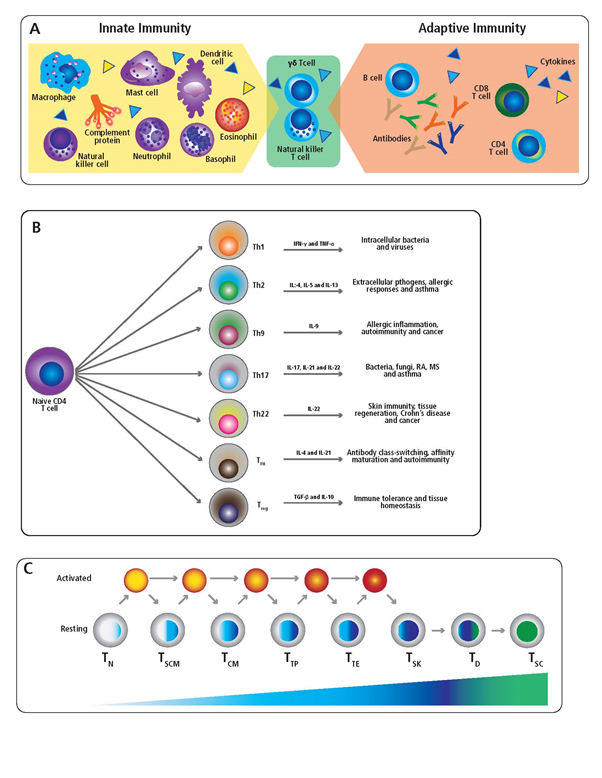
Figure 1: Components of the innate and adaptive immune system. (A) The human immune system is composed of both the innate and adaptive arms. Cells of the innate immune system are indicated in the yellow shaded region, along with innate effector molecules such as cytokines (triangles) and complement. In the green shaded region, specialized T cell populations with innate-like properties, which serve to bridge the gap between innate and adaptive immunity, are highlighted. Components of the adaptive immune system including B cells, secreted antibody, T cells and cytokines are indicated in the orange shaded region. (B) Schematic of CD4+ T cell subsets, including their principal cytokine signature and association with specific immune responses. (C) A model of progressive CD8+ T cell differentiation is illustrated. The CD8+ T cell subpopulations are, as specified in the text: naïve T cells, TN; stem cell-like memory T cells (TSCM); central memory T cells (TCM); polyfunctional memory T cells (TP); Terminal effector T cells (TTE); silent killer effector cells (TSK); dysfunctional T cells (TD); and senescent T cells (TSC). Blue shading of resting T cell populations denotes progressive differentiation and loss of effector functionality. Prolonged activation of CD8+ T cells leading to progressive differentiation, including bypass of developmental intermediates, and transition toward loss of proliferative capacity is depicted using yellow to red shading. Green shading of TD and TSC subpopulations denotes T cell exhaustion and loss of effector function.
The adaptive immune system, consisting of B and T lymphocytes, bridges this gap. B cells are responsible for the production of antibody (immunoglobulin), while T cells engage in direct cell-mediated immunity1. B and T cells are fundamentally different from cells of the innate immune system in as much as they are not limited to recognition of common pathogen features. Instead, individual B or T cells bear highly variable and unique antigen receptors, termed B cell or T cell receptors, respectively3. While any B or T cell is reactive with only a limited number of antigens, collectively all B and T cells in the body can recognize essentially any self- or foreign-antigen. Both B and T cells bestow the adaptive immune system with immunologic memory, that is, the capacity to respond to an antigenic insult with increased speed and vigor upon a secondary encounter4.
The frequency of B or T cells specific for any given antigen is, with few exceptions, very low5, 6. The task of detecting them is therefore akin to finding a “needle in a haystack”. Following a viral or bacterial infection, or as the consequence of vaccination, very rare antigen-specific naive B and T cells (<< 1 in 106 white blood cells, also called peripheral blood mononuclear cells, PBMC) engage in vigorous proliferation, and their frequencies increase sharply7-11. Within 10 days of initiating an immune response, antigen-specific B and T cells can account for up to 1%, but more typically 0.1%, of all lymphocytes in PBMC12-14. In the subsequent weeks, irrespective of whether the antigen is completely cleared or chronically persists, these antigen-specific populations contract and their frequencies decline15. Typically, antigen-experienced memory cell populations occur in the 1:104 to 1:106 frequency range among all B and T cells, and are stably maintained for years, if not decades, even in the absence of antigen re-encounter16, 17.
While standard flow cytometric approaches are well-suited for studying antigen-specific T cells that exist in the 1:103 – 1:104 frequency range, these methods are not ideally suited for detecting memory T cells that exist at lower frequencies18 (Table 1). Yet memory T cells specific for most antigens occur in substantially lower frequencies. ImmunoSpot® assays represent an alternative approach to these traditional flow cytometry methodologies since they are more sensitive and reliably detect antigen-specific memory T cell populations as rare as 1:106 17, 19. The sensitivity of ImmunoSpot® assays is not only essential for the mere identification of antigen-specific memory T cells in peripheral blood, but in particular for segregating these cells into subpopulations to ascribe their distinct effector functions. To this end, immune monitoring aims to establish both the frequency of antigen-specific memory T cells in the blood and their effector profile as a measure of immunity20.
Table 1. Assay platforms for assessment of T cell immunity. Approaches for evaluating T cell immunity, including cytokine production and proliferation, along with advantages and drawbacks are listed.
| Assay | Advantages | Disadvantages |
|---|---|---|
| ELISPOT / FluoroSpot | - Low cell input requirement/assay miniaturization - Single-cell resolution/low limit of detection - Multi-analyte detection/multiplexing - High-throughput scalability - Functional phenotyping - Non-destructive/cell re-utilization |
- No surface phenotype - Limitation in multiplexing - Assay interference due to cytokine capture - Compatible antibody pair requirements - No direct measurement of cell proliferation |
| Tetramer / Multimers | - Direct identification/quantification of peptide-specific T cell populations - Compatible with multi-parametric surface phenotyping - Single-cell resolution - Parallel assessment of independent T cell populations using multiple detection probes |
- Assay sensitivity is dependent on T cell receptor avidity and expression level - Restricted to assessment of predefined T cell populations based on reagent availability - Limited to donors with specific HLA genotypes |
| ELISA | - Simple - Wide range of commercially available kits - High-throughput scalability/batch analysis |
- No ability to multiplex - Lower sensitivity due to consumption of analyte during culture stimulation - Sample volumes limit assessment of multiple analytes - No surface phenotype |
| Proliferation assay (CFSE) | - Direct measure of T cell division - Compatible with multi-parametric surface phenotyping - Single-cell resolution |
- Requires fluorescent labeling of lymphocytes prior to antigen stimulation - High cell input requirements/limit of detection - Limited throughput/scalability |
| Luminex | - Multi-analyte detection/multiplexing - High-throughput scalability/batch analysis |
- No surface phenotype - Poor resolution/limit of detection - Lower sensitivity due to consumption of analyte during culture stimulation |
| ICS | - Multi-analyte detection/Multiplexing - Compatible with multi-parametric surface phenotyping - Single-cell resolution - Functional phenotyping |
- Inhibition of secretion required for analyte detection - High cell input requirements/limit of detection - Limited throughput/scalability - Destructive method |
Antigen-specific T cells belong to either the CD4+ or CD8+ lineage. While CD8+ T cells preferentially recognize antigens that are biosynthesized by infected or transformed host cells, CD4+ T cells typically target protein antigens acquired from the extracellular space1. Upon antigen encounter and appropriate co-stimulatory cues, both CD4+ and CD8+ T cells differentiate into discrete memory cell lineages that secrete distinct and often mutually exclusive sets of cytokines15, 21. Several distinct CD4+ T helper (Th) subsets have been identified, and in particular the Th1, Th2 and Th17 lineages have been most extensively characterized. However, additional CD4+ T cells subsets have gained notoriety in recent years and include the Th9 and Th22 subsets that secrete IL-9 and IL-22 respectively22, 23. Moreover, follicular T cells (Tfh) migrate into B cell follicles due to upregulation of CXCR5 and support germinal center reactions and antibody production through production of IL-2124. Furthermore, FoxP3+ regulatory T cells (Treg) are a specialized CD4+ subset that exhibit immunosuppressive activity25.
In the context of the well-characterized Th1, Th2 and Th17 lineages, Th1 cells are capable of interferon gamma (IFN-γ), but not interleukin 4 (IL-4) or interleukin 17 (IL-17) secretion26, 27. Th1 cells are also important for promoting an inflammatory response that confers protection against most viruses28. Th2 cells secrete IL-4, but not IFN-γ or IL-17, and likely originally evolved to convey anti-parasite immunity29. However, especially in industrialized countries, Th2 cells are more commonly associated with allergies30. Lastly, Th17 cells secrete IL-17, but not IFN-γ or IL-4, and are critical for mounting effective immune responses against intracellular pathogens and fungi31. Autoantigen-reactive Th17 cells are also thought to be the main drivers of autoimmune pathology32.
Whereas secretion of IFN-γ, IL-4 and IL-17 are mutually exclusive to the Th1/Th2/Th17 lineages, respectively, production of IL-9, IL-21 or IL-22 is not necessarily restricted to a single T helper subset. Specifically, IL-9 can be produced by several subsets of CD4+ T cells in addition to Th9 cells, including the Th2, Th17 and Treg lineages33. Moreover, Th9 polarized cells can also produce IL-10 and IL-2123. Th9 cells have been associated with immunity against parasites and tumors, but are also linked to allergic inflammation and autoimmune diseases34. Likewise, IL-21 is produced by several CD4+ T cell subsets, including Th9 and Th17, and therefore is not restricted to the Tfh subset35. Additionally, while expression of IFN-γ, IL-17 or IL-5 is rare amongst human Tfh cells36, these cells are also defined by IL-4 production37. Furthermore, Tfh cells in mouse models can acquire expression of IFN-γ or IL-17 under certain circumstances38, 39. Tfh are closely associated with protective humoral immunity through their ability to promote immunoglobulin class-switching and affinity maturation in B cells. However, increased frequencies of circulating Tfh-like cells also occur in certain autoimmune diseases associated with elevated levels of autoantibody40-43. Lastly, the IL-22 cytokine is produced by both Th22 and Th17 lineages44, 45. Th22 cells are mostly associated with immune responses and tissue regeneration occurring in the skin46. However, Th22 cells and/or IL-22 levels are also negatively associated with Crohn’s diseases47 and cancer48, 49.
Due to the fact that the Th1/Th2/Th17 memory T cell lineages elicit fundamentally different inflammatory response patterns upon antigen re-encounter, it is insufficient for immune diagnostic purposes to enumerate only the frequency of antigen-specific T cells. Instead, it is essential to understand whether the Th1, Th2 or Th17 cell lineage is dominant within the antigen-specific memory T cell pool. By counting the number of individual antigen-specific T cells in PBMC that secrete IFN-γ vs. IL-4 vs. IL-17 following cognate antigen stimulation, immune monitoring seeks to establish the ratio of Th1/Th2/Th17 memory T cells in the blood, and thus to characterize the quality of T cell immunity in individual test subjects.
An important factor that needs to be considered when establishing the ratio of Th1/Th2/Th17 antigen-specific T cells in the blood is that the kinetics of IFN-γ, IL-4 and IL-17 production are very different. IFN-γ production by Th1 cells peaks 24h after antigen encounter, IL-4 secretion by Th2 cells peaks at 48h, and IL-17 production by Th17 cells peaks 3-4 days after antigen stimulation (not even being detectable at 24h/48h)50. As a consequence of these non-overlapping secretion kinetics, Th2 and Th17 antigen-specific T cells are likely to go undetected when immune monitoring is performed at a single time-point, typically within 6-24h following antigen stimulation, as has traditionally been done.
Immune monitoring also aims to establish the life history of memory T cells. Antigen-specific memory T cells’ ability to contribute to host defense is particularly high if these T cells are still in their prime and capable of proliferative expansions51. In contrast, as a consequence of chronic antigen exposure, T cells can be driven to exhaustion and become dysfunctional52. The cytokine secretion profile observed following antigen stimulation therefore enables distinction between these two extremes, as well as intermediate stages of a T cell’s life history (Table 2)51.
In the remainder of this mini-review we will focus on the secretory signature of CD8+ memory T cell subpopulations. While the list of CD8+ T cell subsets continues to expand, and is beyond the scope of this review, the underlying theme remains the same. Namely, naïve CD8+ T cells (TN) lack cytolytic activity and fail to produce cytokines following their initial antigen encounter53. However, within 5 days of antigen receptor engagement, naive CD8+ T cells acquire effector functions and produce high levels of perforin and granzyme B (GzB) enabling them to efficiently kill infected or transformed target cells54. These T cells are now termed terminal effector cells (TTE). They are capable of producing large quantities of GzB, perforin, IFN-γ and TNF-α, but TTE produce little if any IL-255, 56. In the absence of ongoing antigen receptor signaling, activated effector CD8+ T cells return to a quiescent state to become central memory cells (TCM)57. TCM are capable of IFN-γ secretion within 24h of antigen recall but have (temporarily) lost the ability to secrete GzB and perforin. CD8+ TCM cells also secrete little to no TNF-α or IL-2 within the first 24h after antigen re-encounter, but within 3-4 days of antigen stimulation these cells can convert again into TTE56.
Another CD8+ memory T cell subset termed “stem cell-like memory” (TSCM) has gained increasing attention in recent years58. In the context of adoptive cancer therapy, it was observed that transfer of fully differentiated CD8+ T cell effectors resulted in less tumor clearance and worse clinical outcome relative to injection of less-differentiated CD8+ T cells59, 60. TSCM are capable of extensive proliferation following antigen re-encounter, a trait that is progressively lost by other CD8+ memory cell subpopulations following repetitive antigen stimulation. Due to their self-renewing potential and longevity, TSCM can give rise to large numbers of TTE cells following antigen re-encounter, and are therefore especially important when a sustained CD8+ T cell response is required. Following antigen stimulation, TSCM secrete negligible amounts of IFN-γ and TNF-α, and lack expression of GzB. However, TSCM produce abundant quantities of IL-256.
There is also a subpopulation of terminal effector CD8+ T cells, termed “silent CD8+ killer cells” (TSK) that do not secrete IFN-γ, and instead exert their cytolytic effector function through the production of GzB and perforin, without eliciting a local inflammatory response61.
Amongst CD8+ memory T cells there is a subpopulation that co-expresses IFN-γ with IL-2 and TNF-α. These so-called “polyfunctional” T cells (TP) represent a minor percentage of the total antigen-specific CD8+ T cell pool, but their presence in elevated frequencies has been linked to protective immune responses against infectious diseases and cancer62.
CD8+ T cells will undergo exhaustion if repetitively stimulated in the context of a high antigen burden, as frequently occurs in the context of anti-tumor immunity, autoimmunity and chronic infections. “Dysfunctional” CD8+ T cells arise (TD) that have lost their proliferative capacity and exhibit impaired cytolytic activity and reduced cytokine secretion62. While not completely lacking functionality, these cells possess attenuated effector functions to avoid excessive collateral damage in the tissue(s) in which the antigen persists. With ongoing stimulation, these CD8+ T cells will eventually burn out and become “senescent” (TSC). Senescent CD8+ T cells fail to secrete the pro-inflammatory cytokines IFN-γ, TNF-α or IL-2 in response to antigen stimulation. Instead, TSC secrete IL-6 and IL-8, which primarily serves to promote tissue repair but also promotes tumor growth63.
As shown in Table 2, many of the CD8+ T cell subpopulations described above can be identified/segregated based on their secretory signature through measurement of IFN-γ, TNF-α, IL-2 and GzB production following antigen challenge. Since these subpopulations occur in rather low frequencies, four-color ImmunoSpot® assays particularly lend themselves to dissecting the expression/co-expression pattern of these analytes at single-cell resolution64. For the accurate identification of these distinct CD8+ subpopulations, it is critical that each analyte is measured without signal overlap. This is accomplished by using combinations of fluorochromes that can be completely separated, and each color plane analyzed, without the need for compensation, essentially as a single-color ELISPOT assay. Next, images containing the “spots” for the different color planes are superimposed to identify cells that co-express analytes (see Figure 2). This cannot be accomplished by simply matching centers of masses for the individual spots because analyte production is asynchronous and T cells migrate during the assay’s capture phase which lasts 24h or more. Thus, tolerances need to be accommodated as we have described previously65. Moreover, it has been shown that in the low frequency range in which antigen-specific T cells occur, the chance for random overlays of single-positive spots occurring is essentially nonexistent. Once single- vs. multiple-analyte-producing cells are identified, which is done automatically by the ImmunoSpot® software, the data is immediately converted into flow cytometry standard (FCS) format. Using standard software, the various analyte-producing populations can be analyzed for co-expression levels and patterns in a very similar fashion as is traditionally performed following intracellular cytokine staining using a flow cytometer.
Table 2. Dissection of CD8+ memory T cell subpopulations based on analyte secretion profiles. At different stages of their differentiation, the specified CD8+ memory T cell subpopulations secrete distinct patterns of interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 2 (IL-2) and granzyme B (GzB). The subpopulations are, as specified in the text: naïve T cells, TN; stem cell-like memory T cells (TSCM); central memory T cells (TCM); polyfunctional memory T cells (TP); Terminal effector T cells (TTE); silent killer effector cells (TSK); dysfunctional T cells (TD); and senescent T cells (TSC).
| CD8+ T cell Subset | Proliferation | IFN-γ | TNF-α | IL-2 | GzB |
|---|---|---|---|---|---|
| TN | +++ | - | - | - | |
| TSCM | +++ | - | +++ | ||
| TCM | ++ | +++ | +/- | +/- | - |
| TP | +++ | +++ | ++ | - | |
| TTE | +++ | +++ | - | +++ | |
| TSK | - | +++ | |||
| TD | +/- | +/- | - | - | |
| TSC | - | - |
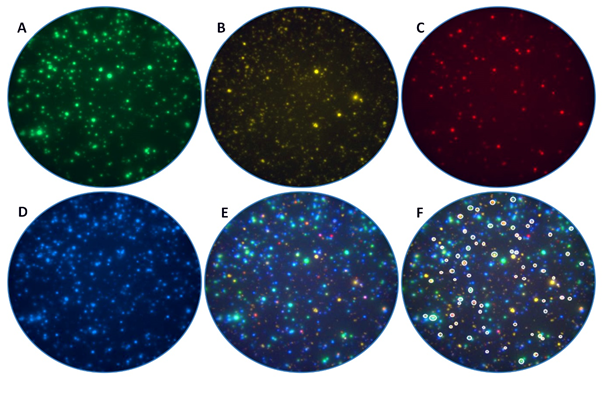
Figure 2: Dissection of CD8+ T cell subpopulations using a four-color ImmunoSpot® assay
Human peripheral blood mononuclear cells were stimulated with antigen and the elicited interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 2 (IL-2) and/or granzyme B (GzB) captured on the membrane around the individual T cells that secreted the respective analyte(s). The individual analytes are selectively detected in the (A) IFN-γ, (B) TNF-α, (C) IL-2 and (D) GzB channels, respectively. Note the lack of signal cross-bleeding, which permits identification of each analyte in its own color plane without the need for compensation. (E) Composite overlay of the four individual channels, enable identification of analyte co-expression patterns by individual CD8+ cells. (F) Polyfunctional CD8+ T cells (TP) that secrete IFN-γ, TNF-α, IL-2 and GzB are highlighted using white outlining.
In conclusion, multi-color ImmunoSpot® assays are ideally suited for assessing rare populations of antigen-experienced T cells and enable segregation of functionally distinct T cell lineages on the basis of their secretory signatures. Additionally, taking into account the assay’s ease of implementation, efficient utilization of white blood cells, high-throughput capability, automated evaluation, as well as audit trails generated for regulatory compliance, multi-color ImmunoSpot® assays have great potential to revolutionize the immune monitoring field through providing detailed information on the magnitude, quality, and antigen life history of T cells.
Conflict of Interest Statement: PVL is the founder and CEO of Cellular Technology Limited (CTL), a company that specializes in ELISPOT testing, producing high-throughput-suitable readers, test kits, and GLP-compliant contract research. GAK, JH and DRR are employees of CTL
References
- Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010; 125(2 Suppl 2): S3-23.
- Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009; 22(2): 240-73, Table of Contents.
- Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010; 125(2 Suppl 2): S33-40.
- Gourley TS. Generation and maintenance of immunological memory. Semin Immunol. 2004; 16(5): 323-33.
- Blattman JN, Antia R, Sourdive DJ, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002; 195(5): 657-64.
- Pape KA, Taylor JJ, Maul RW, et al. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011; 331(6021): 1203-7.
- Pape KA, Taylor JJ, Maul RW, et al. Induction of human plasmablasts during infection with antibiotic-resistant nosocomial bacteria. J Antimicrob Chemother. 2014; 69(7): p. 1830-3.
- Callan MF, Tan L, Annels N, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J Exp Med. 1998; 187(9): 1395-402.
- Sakabe S, Sullivan BM, Hartnett JN, et al. Analysis of CD8(+) T cell response during the 2013-2016 Ebola epidemic in West Africa. Proc Natl Acad Sci U S A. 2018; 115(32): E7578-E7586.
- Subbramanian RA, Basha S, Shata MT, et al. Pandemic and seasonal H1N1 influenza hemagglutinin-specific T cell responses elicited by seasonal influenza vaccination. Vaccine. 2010; 28(52): 8258-67.
- Wrammert J, Onlamoon N, Akondy RS, et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol. 2012; 86(6): 2911-8.
- Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 1998; 8(2): 167-75.
- Ellebedy AH, Jackson KJ, Kissick HT, et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol. 2016; 17(10): 1226-34.
- Masopust D, Murali-Krishna K, Ahmed R. Quantitating the magnitude of the lymphocytic choriomeningitis virus-specific CD8 T-cell response: it is even bigger than we thought. J Virol. 2007; 81(4): 2002-11.
- Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007; 27(3): 393-405.
- Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014; 14(1): 24-35.
- Helms T, Boehm BO, Asaad RJ, et al. Direct visualization of cytokine-producing recall antigen-specific CD4 memory T cells in healthy individuals and HIV patients. J Immunol. 2000; 164(7): 3723-32.
- Bacher P, Scheffold A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A. 2013; 83(8): 692-701.
- Barabas S, Spindler T, Kiener R, et al. An optimized IFN-gamma ELISpot assay for the sensitive and standardized monitoring of CMV protein-reactive effector cells of cell-mediated immunity. BMC Immunol. 2017; 18(1): 14.
- Keilholz U, Martus P, Scheibenbogen C. Immune monitoring of T-cell responses in cancer vaccine development. Clin Cancer Res. 2006; 12(7 Pt 2): 2346s-2352s.
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010; 28: 445-89.
- Azizi G, Yazdani R, Mirshafiey A. Th22 cells in autoimmunity: a review of current knowledge. Eur Ann Allergy Clin Immunol. 2015; 47(4): 108-17.
- Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015; 15(5): 295-307.
- Vinuesa CG. Follicular Helper T Cells. Annu Rev Immunol. 2016; 34: 335-68.
- Sakaguchi S, Miyara M, Costantino CM, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010; 10(7): 490-500.
- Karulin AY, Hesse MD, Tary-Lehmann M, et al. Single-cytokine-producing CD4 memory cells predominate in type 1 and type 2 immunity. J Immunol. 2000; 164(4): 1862-72.
- Kuerten S, Rottlaender A, Rodi M, et al. The clinical course of EAE is reflected by the dynamics of the neuroantigen-specific T cell compartment in the blood. Clin Immunol. 2010. 137(3): 422-32.
- Kaiko GE, Horvat JC, Beagley KW, et al. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008; 123(3): 326-38.
- Choi P, Reiser H. IL-4: role in disease and regulation of production. Clin Exp Immunol. 1998; 113(3): 317-9.
- Georas SN, Guo J, De Fanis U, et al. T-helper cell type-2 regulation in allergic disease. Eur Respir J. 2005; 26(6): 1119-37.
- Guglani L Khader SA. Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS. 2010; 5(2): 120-7.
- Tabarkiewicz J, Pogoda K, Karczmarczyk A, et al. The Role of IL-17 and Th17 Lymphocytes in Autoimmune Diseases. Arch Immunol Ther Exp (Warsz). 2015; 63(6): 435-49.
- Tan C, Gery I. The unique features of Th9 cells and their products. Crit Rev Immunol. 2012; 32(1): 1-10.
- Li J, Chen S, Xiao X, et al. IL-9 and Th9 cells in health and diseases-From tolerance to immunopathology. Cytokine Growth Factor Rev. 2017; 37: 47-55.
- Yi JS, Cox MA, Zajac AJ. Interleukin-21: a multifunctional regulator of immunity to infections. Microbes Infect. 2010; 12(14-15): 1111-9.
- Kroenke MA, Eto D, Locci M, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol. 2012; 188(8): 3734-44.
- Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011; 29: 621-63.
- Ding Y, Li J, Wu Q, et al. IL-17RA is essential for optimal localization of follicular Th cells in the germinal center light zone to promote autoantibody-producing B cells. J Immunol. 2013; 191(4): 1614-24.
- Yusuf I, Kageyama R, Monticelli L, et al. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol. 2010; 185(1): 190-202.
- He J, Tsai LM, Leong YA, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013; 39(4): 770-81.
- Morita R, Schmitt N, Bentebibel SE, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011; 34(1): 108-21.
- Simpson N, Gatenby PA, Wilson A, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010; 62(1): 234-44.
- Szabo K, Papp G, Barath S, et al. Follicular helper T cells may play an important role in the severity of primary Sjogren's syndrome. Clin Immunol. 2013; 147(2): 95-104.
- Kreymborg K, Etzensperger R, Dumoutier L, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. 2007; 179(12): 8098-104.
- Duhen T, Geiger R, Jarrossay D, et al. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009; 10(8): 857-63.
- Fujita H. The role of IL-22 and Th22 cells in human skin diseases. J Dermatol Sci. 2013; 72(1): 3-8.
- Brand S, Beigel F, Olszak T, et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006; 290(4): G827-38.
- Qin S, Ma S, Huang X, et al. Th22 cells are associated with hepatocellular carcinoma development and progression. Chin J Cancer Res. 2014; 26(2): 135-41.
- Yang XW, Jiang HX, Lei R, et al. Recruitment and significance of Th22 cells and Th17 cells in malignant ascites. Oncol Lett. 2018; 16(4): 5389-5397.
- Duechting A, Przybyla A, Kuerten S, et al. Delayed Activation Kinetics of Th2- and Th17 Cells Compared to Th1 Cells. Cells. 2017; 6(3).
- Martin MD, Badovinac VP. Defining Memory CD8 T Cell. Front Immunol. 2018; 9: 2692.
- Hashimoto M, Kamphorst AO, Im SJ, et al. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med. 2018; 69: 301-318.
- Brenchley JM, Douek DC, Ambrozak DR, et al. Expansion of activated human naive T-cells precedes effector function. Clin Exp Immunol. 2002; 130(3): 432-40.
- Golstein P, Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev Immunol. 2018; 18(8): 527-535.
- Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. 2008; 180(3): 1309-15.
- Mahnke YD, Brodie TM, Sallusto F, et al. The who's who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. 2013; 43(11): 2797-809.
- Abdelsamed HA, Moustaki A, Fan Y, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017; 214(6): 1593-1606.
- Lugli E, Dominguez MH, Gattinoni L, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. 2013; 123(2): 594-9.
- Flynn JK, Gorry PR. T cell therapies-are T memory stem cells the answer? Ann Transl Med. 2015; 3(17): 251.
- Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005; 115(6): 1616-26.
- Schlingmann TR, Shive CL, Targoni OS, et al. Increased per cell IFN-gamma productivity indicates recent in vivo activation of T cells. Cell Immunol. 2009; 258(2): 131-7.
- Apetoh L, Smyth MJ, Drake CG, et al. Consensus nomenclature for CD8(+) T cell phenotypes in cancer. Oncoimmunology. 2015; 4(4): e998538.
- Naugler WE, Karin M. The wolf in sheep's clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008; 14(3): 109-19.
- Hanson J, Roen DR, Lehmann PV. Four Color ImmunoSpot((R)) Assays for Identification of Effector T-Cell Lineages. Methods Mol Biol. 2018; 1808: 51-62.
- Karulin AY, Megyesi Z, Caspell R et al. Multiplexing T- and B-Cell FLUOROSPOT Assays: Experimental Validation of the Multi-Color ImmunoSpot((R)) Software Based on Center of Mass Distance Algorithm. Methods Mol Biol. 2018; 1808: 95-113.