
Development, maintenance and functions of CD8+ T-regulatory cells: Molecular orchestration of FOXP3 transcription
Sreeparna Chakraborty1 & Gaurisankar Sa1*
1Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
Abstract
Modulation of immune cells to rejuvenate the immune responses against cancer becomes a promising strategy for cancer therapy. T-regulatory cells are one of the major hurdles in successful cancer immunotherapy. Recent studies discovered that apart from CD4+ Treg cells, CD8+ Tregs also play roles in tumor immune evasion. Moreover, CD8+ Tregs shows synergistic immunosuppression with CD4+ Treg cells in tumor microenvironment. Several phenotypic markers have been described for peripherally induced CD8+ Treg cells, but till now no universal phenotypic signature has yet established. FOXP3 is the master regulator of Treg cells and its transcription is critically regulated by promoter region as well as three intronic conserved non-coding regions, viz; CNS 1, 2 and 3. In this review, we have described the transcriptional networking associated with the regulation of FOXP3 in tumor-CD8+ Treg cells along with CD4+ nTreg and iTreg cells. Intervention of the intensive transcriptional machinery of FOXP3 regulation may aid to target Treg cells and thus could potentiate immunotherapy of cancer.
Introduction
A new tide in cancer therapy encourages the immunotherapy to become a clinically potential way to fight against this deadly disease. In recent advances several immunotherapeutic strategies are evolving which include cancer vaccines, adoptive transfer of ex vivo activated T and natural killer cells, CART cell and administration of antibodies or recombinant proteins that could block the immune checkpoint pathways i.e. co-stimulatory molecules1. The tumor microenvironment is enriched with cancer-associated fibroblasts, myeloid-derived suppressor cells, and T- regulatory (Treg) cells. In normal physiology Treg cells are potent suppressors of effector T cells and thereby act as a self-check mechanism to maintain the peripheral tolerance and homeostatic in immune response. However, expanded densities of tumor-associated Treg cells have been related to poor prognosis in various cancers1. The predominance of Treg cells in the tumor microenvironment is found to be one of the main impediments to successful cancer immunotherapy. Therefore, one methodology for initiating compelling hostile to tumor insusceptibility is to selective depletion of differentiated Treg along with the activation of tumor-specific effector T cells by using cancer vaccine or immune checkpoint blockade and these might unleash potent antitumor responses to make the current cancer immunotherapy more effective. Till now the involvement of FOXP3-positive CD4+ Treg cells with the advancement of cancer is well established2, 3 . However, the interest in the role of CD8+ Treg cells in tumor immunomodulation was resurrected, in part, by the occurrence of this suppressor cells in the tumor microenvironment along with CD4+ Treg cells.
CD8+ Treg cell: a new recruitment to T-regulatory cell family
In the 1970s Grehson et al. first reported the existence of CD8+ Treg cells4. Tregs are generally classified as either ‘natural’ or nTreg or ’inducible or iTreg’. The nTregs develop in the thymus and helps to maintain the homeostasis, while iTregs arise in the periphery following stimulation from microenvironment. The nCD8+ Tregs share functional and phenotypic similarities with CD4+ Tregs and express CTLA4, GITR and FOXP35. However, for peripherally induced CD8+ Treg cells, till now no universal phenotypic emblem has yet established. The involvement of CD8+ Treg cells have been discussed in many autoimmune diseases, inflammatory diseases as well as in transplantation biology6. The predominance of suppressor CD8+CD28−CD56+ cells, CD8+CD11c+ cells and CD8+CD25+Foxp3+ Tregs in RA (rheumatoid arthritis)7-9, CD8+CD122+ and CD8+LAP+( latency-associated-peptide) Treg in EAE (Experimental- autoimmune-encephalomyelitis)9,10, CD8+CD28− T cells in Myasthenia gravis is reported11. In multiple sclerosis the predominance of CD8+CD25+FOXP3+ T cells is profound12. Moreover, the role of CD8+Foxp3+ Treg in GVHD (Graft-versus-host disease)13 and CD8+LAG-3+(Lymphocyte-activation-gene-3)CD25+FoxP3+CCL4 (Chemokine-ligand-4)+14 Treg in inflammatory infectious diseases is already documented. Recent advances in research suggest the prevalence of CD8+CCR7(C-C chemokine-receptor-type-7 )+IL10+ cells in ovarian cancer which suppress the anti-tumor immune response via TGFβ1 and IFNg production15 and CD8+CD28− cells in non-small cell lung carcinoma16. On the other hand, in prostate cancer, CD8+CD25+CD122+FOXP3+GITR(glucocorticoid-induced TNFR-related protein)+ show their suppressive functions via Toll-like receptor-8 (TLR8) signaling17 whereas CD8+CD25+FOXP3+ cells in colon cancer that inhibit CD4+ T cell proliferation and Th1 cytokine production18. Our research reported the occurrence of CD8+CD25+FOXP3+ Treg cells in breast tumor microenvironment19. Further study identified that this subset is CTLA4+/PD-1+/CD127- in nature19. These CD8+ Treg cells secrete IL10 and TGFβ in the tumor microenvironment to suppress the effector T cells proliferation and functions. Moreover, it was observed that CD8+ Treg showed synergistic immunosuppression along with CD4+ Treg cells which further emphasize their importance in tumor immune evasion19.
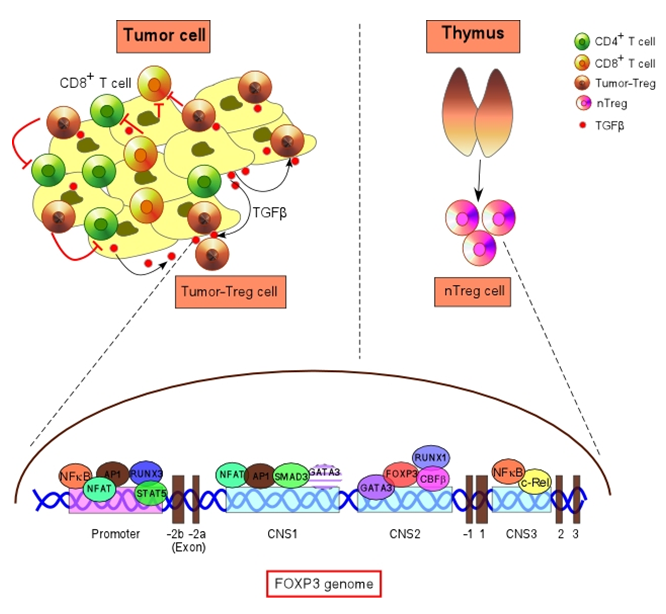
Figure : The transcription of FOXP3 in nTreg and tumor-Treg: an integrative model.
Expression of the FOXP3 gene is controlled by promoter and several regulatory regions. Several transcription factors viz; RUNX3, NFAT, AP1, STAT5 associated with FOXP3-promoter activity. Three conserved non-coding sequences (CNS1, CNS2 and CNS3) have been identified. NFAT, AP1 and SMAD3 bind to CNS1, which are required for chromatin modification and FOXP3 induction during tumor-Treg cell differentiation. CNS2 region associated with FOXP3 maintainence by GATA3, RUNX1–CBFβ complex and FOXP3. CNS3 region activity asgined to nTreg cells by binding of NFκB and c-Rel.
FOXP3: the ace controller of Treg cell
The FOXP3 gene is found on the X chromosome and mutation or loss of function of FOXP3 leads to severe autoimmune or inflammatory diseases. Research established that FOXP3 is the master regulator of Treg cells and the stable FOXP3 expression is critical for proper differentiation and immunosuppressive function of Treg cells20.
FOXP3 acts as a transcription regulator and switches on a complex transcriptional network that ensures the Treg phenotype and functions. In one hand, FOXP3 acts as a transcriptional activator to induce the expression of CD25 (IL2-Rα) and CTLA4 (negative Co-stimulatory molecule)21. On the other hand, it acts as a repressor to inhibit IL17 transcription by binding to RORg or FOXP3 competes with AP-1 for cooperative binding with NFAT and binds on the IL2 promoter to repress IL2 transcription21, 22.
FOXP3 promoter is tightly regulated by a number of transcription factors
The human FOXP3 promoter is stretched from -6221 bp upstream of the TSS but is interrupted by a 6000-bp intron at 5’ UTR23. The eukaryotic promoter contains several consensus sequences viz; TATAAA sequence known as TATA box, located at -30 to -25 bp upstream of the TSS, GGGCGG sequence i.e. GC box located at 138bp upstream of the TSS and CAAT box contains GGCCAATCT consensus sequence (218bp upstream of the TSS). Likewise in the human FOXP3 genome, TATA box is situated at -211/ 176 bp whereas GC box is at -307/ 176 bp and have specific transcription factors binding motif23. In thymus-derived nTreg cells, TCR-mediated signaling plays critical roles in FOXP3 transcription initiation. NFκB, NFAT, and AP1 are well-recognized transcription factors that are induced following TCR engagement and ensure the T cell activation23-25. FOXP3 promoter contains three NFAT and AP1 binding sites which play definitive roles in the trans-activation of FOXP323. NFκB also has potential binding in the putative cis-elemen in the FOXP3 promoter region and induce FOXP3 transcription in nTreg cells24. Moreover, IL2 activated STAT5 binds to the FOXP3 promoter and acts as a positive trans-activator of FOXP326. Human FOXP3 promoter also contains three conserved putative RUNX3 responsive elements (at −720 bp, −670 bp and −441 bp upstream of the TSS). RUNX3 plays a decisive role in the differentiation of CD8+ T cells from CD4/CD8-double positive progenitor cells27. However, in tumor-induced CD8+ Treg cells this runt domain transcription factor, RUNX3, acts as positive trans-activator of FOXP3 by binding at the putative site in the promoter region and causes permissive chromatin modification19.
FOXP3 induction and maintenance is regulated by conserved non-coding sequences
Apart from the promoter region, alignment of the FOXP3 gene from different species has revealed the presence of a few monitored conserved non-coding regions viz; CNS1, CNS2, and CNS3. These conserved non-coding DNA sequence (CNS) elements encode information that defines the differentiation and stability of the Treg cells. The CNS1 region is exclusively associated with TGFβ-induced FOXP3 induction in peripherally induced Treg cells, whereas CNS3 activity is profound in thymus-derived nTreg cells. Upon TCR stimulation NFκB and c-Rel bind to the CNS3-responsive elements and induce FOXP3 transcription. On the other hand, the CNS2 region is exclusively associated with the maintenance of FOXP3 expression in both nTreg and iTreg cells28.
Research showed that CNS1 region is profoundly associated with tumor-induced CD8+ as well as CD4+ iTreg generation. The CNS1 region is extended from −372 bp upstream of the transcription start site to +1746 bp of the human FOXP3 gene locus. It contains TGFβ responsive element-SMAD3 (at +1544 bp region) which is essential for peripheral induction of FOXP3 expression in iTreg cells19, 25. Transforming growth factor-β (TGF-β) is a polypeptide growth factor which regulates divergent cellular processes like proliferation, differentiation, and apoptosis29. TGF-β applies a multifaceted impact on malignancy pathogenesis and regulates the insensitivity to antigrowth signals or evasion of apoptosis. It induces the tumor angiogenesis, invasion, and metastasis. TGF-β is abundantly expressed in most of the human tumors and binds to TGFβ-R1 which further triggers the activation of SMAD3 by phosphorylation at the C-terminal serine residues30. The functions of SMAD3 in promoting EMT is well studied31, however, it has a profound role in immune system also. It is observed that tumor-secreted TGFβ acts on responder T cells and causes phosphorylation of SMAD3 which further translocate to nucleus3, 19. SMAD3 binds to the responsive element in CNS1 region of FOXP3 gene and causes permissive histone acetylation in H4 that leads to the induction of FOXP3 transcription in CD8+ Treg cells19. Thus, in TGFβ-induced Treg cells SMAD3 plays an indispensable role in FOXP3 induction.
Bi-functional role of GATA3 in transcription of FOXP3
GATA3 is a member of a GATA-binding protein family that interacts with GAATA motif. GATA3 regulates a variety of biological processes viz; development, differentiation, and tumorigenesis32, 33. Moreover, GATA-3 is critically involved in immune regulation. GATA-3 is known as the ‘‘master regulator’’ for Th2 cell differentiation34. However, the GATA3 function is also constitutively expressed in CD8+ T cells also and very crucial to maintain their functions35. It is reported that Th2 cell polarizing condition antagonizes TGFβ-induced Foxp3 expression and promotes the Th9 cell differentiation36. In silico analysis showed that the CNS1 region in FOXP3 gene contains a conserved GATA3-binding site at +1448 bp region19. On contrary to SMAD3, GATA3 binds on the specific CNS1-responsive element and represses the FOXP3 transcription in tumor-CD8+ Treg cells. Thus, SMAD3 and GATA3 play opposing roles in tumor-induced FOXP3 induction in CD8+ Treg cells via regulating the CNS1-genomic locus.
The theme of Treg cell stability and plasticity has been debatable for a long time. Scientists have found that some FOXP3+CD4+ T cells may lose FOXP3 expression and acquire effector Th cell function depending on their microenvironment37. Our research shows that tumor-induced CD8+ Treg cells are with stable FOXP3 expression over time which certainly indicates its distinctive transcriptional or epigenetic regulation. The CNS2 gene locus is known to be very crucial for sustaining a stable FOXP3 expression in Treg cells28. In association with Cbfβ–Runx1, FOXP3 binds to CNS2 and helps to maintain stable expression of FOXP3 via auto-regulation38. However, the recruitment of FOXP3–Runx1–Cbfβ complexes to CNS2 occurs after and is dependent on demethylation of the CpG island located at this region38. GATA3 induces DNA demethylation in Th2 gene locus and play a critical role in Th2 cell differentiation39. Apart from CNS1, CNS2 region also contains a GATA3-responsive element at +3488 bp19. In tumor-induced CD8+ Treg cells, GATA3 binds to the CNS2-responsive site and helps to maintain stable FOXP3 expression19. Thus, it seems probable that GATA3 helps in maintenance of an active state of the FOXP3 via inducing DNA-demethylation in CNS2 and further helps in the FOXP3–Runx1–Cbfβ mediated auto-regulation of FOXP3.
Conclusion:
As illustrated in this review, Treg cells are a heterogeneous population and their stability and plasticity depend on their microenvironment. It has become clearer with the time that CD4+ and CD8+ Treg cells have overlapping but ndependen differentiation mechanisms in different cancers. A crosstalk between CD4+ Tregs and CD8+ Tregs might play critical roles in tumor immune evasion which certainly indicates the necessity to acquire knowledge on both the Treg cells subsets. With the accumulation of acquaintance on the molecular mechanism of Treg cell generation and their immunosuppressive functions, we expect that in the near future, the hurdles of successful cancer immunotherapy could be achieved.
Acknowledgement
This work was supported by the grants from University grant commission and Department of Science and Technology, Govt. of India.
References
- Finotello F, Trajanoski Z. New strategies for cancer immunotherapy: targeting regulatory T cells. Genome Med. 2017; 9: 10 .
- Hossain DM. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013; 39: 1057-1069.
- Hossain DM. MEK inhibition prevents tumour-shed transforming growth factor-beta-induced T-regulatory cell augmentation in tumour milieu. Immunology. 2015; 144: 561-573.
- Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology. 1970; 18: 723-737.
- Cosmi L. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood. 2003; 102: 4107-4114.
- Dinesh RK, Skaggs BJ, La Cava A, et al. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun Rev. 2010; 9: 560-568.
- Davila E. Cell-based immunotherapy with suppressor CD8+ T cells in rheumatoid arthritis. J Immunol. 2005; 174: 7292-7301
- Seo SK. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004; 10: 1088-1094.
- Notley CA, McCann FE, Inglis JJ, et al. ANTI-CD3 therapy expands the numbers of CD4+ and CD8+ Treg cells and induces sustained amelioration of collagen-induced arthritis. Arthritis Rheum. 2010; 62: 171-178.
- Chen ML, Yan BS, Kozoriz D, et al. Novel CD8+ Treg suppress EAE by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol. 2009; 39: 3423-3435.
- Ben-David H, Sharabi A, Dayan M, et al. The role of CD8+CD28 regulatory cells in suppressing myasthenia gravis-associated responses by a dual altered peptide ligand. Proc Natl Acad Sci U S A. 2007; 104: 17459-17464
- Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010; 67: 625-638.
- Beres AJ. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol. 2012; 189: 464-474.
- Joosten SA. Identification of a human CD8+ regulatory T cell subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci U S A. 2007; 104: 8029-8034.
- Zhang S. Analysis of CD8+ Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol. 2015; 12: 580-591.
- Chen C. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int Immunopharmacol. 2014; 18: 255-261.
- Kiniwa Y. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007; 13: 6947-6958.
- Chaput N. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009; 58: 520-529.
- Chakraborty S. Transcriptional regulation of FOXP3 requires integrated activation of both promoter and CNS regions in tumor-induced CD8(+) Treg cells. Sci Rep. 2017; 7: 1628.
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003; 4: 330-336.
- Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev. 2011; 241: 260-268.
- Wu Y. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006; 126: 375-387.
- Mantel PY. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006; 176: 3593-3602.
- Long M, Park SG, Strickland I, et al. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009; 31: 921-931.
- Tone Y. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008; 9: 194-202.
- Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009; 30: 616-625.
- Woolf E. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc Natl Acad Sci U S A. 2003; 100: 7731-7736.
- Zheng Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010; 463: 808-812.
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103: 295-309.
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005; 19: 2783-2810.
- Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. 2007; 17: 281-293.
- Pandolfi PP. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet. 1995; 11: 40-44.
- Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010; 222: 42-49.
- Zhu J. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004; 5: 1157-1165.
- Wang Y. GATA-3 controls the maintenance and proliferation of T cells downstream of TCR and cytokine signaling. Nat Immunol. 2013; 14: 714-722.
- Dardalhon V. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008; 9: 1347-1355.
- Hori S. Developmental plasticity of Foxp3+ regulatory T cells. Curr Opin Immunol. 2010; 22: 575-582.
- Kitoh A. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity. 2009; 31: 609-620.
- Bird JJ. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998; 9: 229-237.