
Experimental Autoimmune Encephalomyelitis Animal Models Induced by Different Myelin Antigens Exhibit Differential Pharmacologic Responses to Anti-Inflammatory Drugs
Yuxi Yan1#, Quan Zhao1#, Ya Huang1, Janine Y. Yang2, Jie Zou1, Chunxia Ao1, Xiaojuan Chai1, Renhong Tang1 and WenQing Yang1*
1State Key Laboratory of Translational Medicine and Innovative Drug Development, Jiangsu Simcere Pharmaceutical Co., Ltd., Nanjing, Jiangsu, China.
2Massachusetts Eye and Ear, Harvard Medical School, USA
Abstract
Background and objective
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used model for studying autoimmune-mediated myelin degradation in multiple sclerosis (MS). Here, we evaluated the pharmacologic responses of several anti-inflammatory drugs with varying mechanisms of actions (MOAs) using EAE models induced by different MOG immunogens to reveal differential pharmacologic characteristics of the disease models and provide a general guidance in animal model selection for MS research.
Methods
The pharmacologic responses of anti-inflammatory drugs with different mechanisms of actions (MOAs) were evaluated using EAE models induced by either myelin oligodendrocyte glycoprotein p35-55 (MOG35-55) or p1-128 (MOG1-128). EAE animal models were developed in mice with C57BL/6 background. The animals were treated with different anti-MS medications, including 3 B cell-mediated agents and 2 T cell-mediated agents, respectively. Clinical symptoms were monitored and scored, and pharmacodynamic markers including cytokine secretion, inflammatory cell infiltration, and demyelination in spinal cord were analyzed.
Results
In MOG35-55 peptide-induced EAE model, T cell modulating agents Secukinumab and Fingolimod significantly alleviated clinical symptoms, while B cell-depleting agents, BTK inhibitors PRN2246 and Telitacicept, displayed minimal therapeutic effects or even exacerbated disease progression. In contrast, both T cell-modulating agents and B cell-depleting agents ameliorated disease severity in MOG1-128-induced EAE model. T cell and B cell infiltration in spinal cord increased with disease progression in MOG1-128-induced EAE model.
Conclusions
Our results demonstrated that induction of EAE by different myelin antigens resulted in differential pharmacologic responses to drugs with specific MOAs. The MOG35-55 peptide-induced EAE model only responded to T cell-modulating drugs, whereas the MOG1-128 protein-induced EAE model exhibited therapeutic sensitivity to both T cell- and B cell-modulating agents. These data suggest the MOG35-55 peptide-induced EAE model is suitable for assessing T cell-modulating agents while MOG1-128 protein-induced model can be employed to evaluate both T cell- and B cell-modulating agents.
Introduction
Multiple sclerosis (MS) is an autoimmune and neurological disease characterized by demyelination of the central nervous system (CNS). Due to the long course of the disease and high disability rate, MS is a huge socio-economic burden1. Although a few agents, including kinase inhibitors and monoclonal antibodies, have shown promising results in the clinic, MS remains one of the most common causes of neurological dysfunction, highlighting the need to develop novel therapies for MS patients2.
The experimental allergic encephalomyelitis (EAE) mouse model has been widely used in studying the mechanisms of autoimmune-mediated myelin degradation and testing new therapies for MS3. Two commonly used animal models of myelin oligodendrocyte glycoprotein (MOG) antigen-induced EAE include MOG35-55 peptide- and MOG1-125 (or MOG1–128) protein-induced models4. Though both MOG35–55 and MOG1–128 can induce similar clinical symptoms in mice, there are different immunological mechanisms for disease occurrence and progression between these two models. The short peptide MOG35–55 is the dominant MHC II class restricted epitope of myelin proteins, and its pathogenic mechanism for EAE is largely mediated by CD4+ T help (Th) cells. The extracellular domain of MOG (MOG1–128) contains conformational epitopes that can be recognized by B cell receptor (BCR)4-6. As for MOG1-128-induced EAE model, both antigen-activated B cells and antibodies against MOG protein are involved in the pathogenic processes due to the recognition of conformational epitopes on MOG1-128 protein by BCR.
In pharmacological studies, choosing appropriate animal models based on specific MOAs of test agents is a critical process as this may influence data interpretation, especially for a disease model like EAE where the pathogenesis and pathophysiology are complex7. It has been suggested that both T cell- and B cell-mediated inflammatory responses contribute to the initiation and progression of EAE8. However, to our knowledge, there has not been a direct comparison or validation published to systemically investigate the pharmacological effects of commonly used anti-inflammatory drugs in these models.
Food and Drug Administration (FDA)-approved drugs and agents under investigation for MS include corticosteroids, immune-modulating agents, neuroprotective agents, and agents that help improve neuromuscular/synaptic transmission. Based on MOA, immune-modulating agents can be roughly classified into two subsets, B cell modulating/depleting agents and T cell modulating molecules. B cell-acting drugs include Bruton’s tyrosine kinase inhibitors (BTKi) PRN22469 and Evobrutinib10, and the human recombinant fusion protein Telitacicept11. T cell-modulating agents include Secukinumab (an anti-human interleukin-17 biologic12) and Fingolimod (a sphingosine-1-phosphate receptor modulator13), which sequester lymphocytes in the lymph nodes, preventing them from contributing to an autoimmune reaction.
In the present study, we evaluated the pharmacologic responses of several anti-inflammatory drugs with different MOAs using animal models of EAE induced by MOG35-55 or MOG1-128. The results of this study may shed light on the biology of EAE models and guide EAE animal model selection for novel drug candidate profiling or mechanistic investigations in MS research.
Methods and materials
Mice
6- to 8-week-old female C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. C57BL/6-Il17aem2(hIL17A)Il17fem2(hIL17F)Smoc (hIL17A/hIL17F, Cat No: NM-HU-200281) mice were purchased from Shanghai Model Organisms Center, Inc. Animals were housed in a specific pathogen-free barrier condition at OBiO Technology (Shanghai) Corp., Ltd. Animal welfare and experimental procedures were carried out strictly in accordance with animal care and protocol approved by the Institutional Animal Care and Use Committee (IACUC) at OBiO Technology (Shanghai) Corp., Ltd.
Reagents and proteins
PRN2246, Evobrutinib, and Fingolimod were purchased from MedChemExpress Co., Ltd (Princeton, NJ, USA). Secukinumab and Telitacicept were synthesized by Simcere Pharmaceutical (Nanjing, China). MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK; Cat. No.: T510219) was purchased from Sangon Biotech (Shanghai) Co., Ltd. MOG1-128 protein was developed by genetically synthesizing the extracellular domain (aa1-128) of mouse MOG based on amino acid sequence from Uniprot database, cloning it into mammalian expression vector with His-tag expressed in Expi293 cells via transient transfection, and purifying using HisTrap excel column (GE).
Model establishment and in vivo efficacy studies with EAE models
For MOG35-55 peptide-induced EAE model, 6- to 8-week-old female mice were anesthetized with isoflurane and immunized subcutaneously in three sites on the back with a total of 150 μg MOG completely emulsified with equal volume of Incomplete Freund's Adjuvant (Sigma-Aldrich) containing 2.5mg/mL of Mycobacterium tuberculosis H37 Ra (BD DIFCO). This was compared to the immunization agent in MOG1-128 protein-induced EAE model with 225 μg MOG emulsified with Complete Freund's Adjuvant (Sigma-Aldrich). Simultaneously, the mice were administered 200 ng of pertussis toxin intraperitoneally and again at 48 hours after the immunization. The severity of EAE features on these mice was monitored and scored daily for 28 days according to the following standard: 0, no clinical signs; 1, mild tail paralysis or hind limb weakness; 2, tail paralysis and hind limb weakness; 3, partially hind limb paralysis; 4, complete hind limb paralysis and front limb weakness; and 5, near death or death3. Mice were randomly divided into vehicle control or drug treatment groups. For treatment, Fingolimod, PRN2246, and Evobrutinib were administered intragastrically at 0.5, 5, and 10mg/kg/d, respectively. Secukinumab and Telitacicept were diluted in PBS and administered intraperitoneally at 15mpk and 7.5mpk (MOG35-55 induced EAE) and 10mpk and 5mpk (MOG1-121 induced EAE) every 3 days from day 0.
Histopathology and immunohistochemistry (IHC)
Mice were anesthetized and sacrificed at experimental endpoints, and spinal cords were collected and fixed in 10% formalin at the end of the experiment. The cervical, thoracic, and lumbar regions of the spinal cord were separated and embedded in paraffin transversely for four-micrometer histological section, and the rehydrated sections were then stained by Luxol fast blue as well as hematoxylin and eosin (H&E) according to previously described protocols14. The severity of inflammatory cell infiltration and demyelination in stained spinal cord sections was evaluated in double-blinded manner as follows: 0, no cell infiltration or inflammation; 1, little demyelination with few infiltrated cells around the perivascular and meninges area; 2, less than one third part of the white matter is infiltrated by inflammatory cells, and demyelination appears in a few areas; 3, more than one third part of the white matter is infiltrated by inflammatory cells with large areas of demyelination; and 4, the whole white matter is infiltrated by inflammatory cells15. IHC staining of CD4+ T cells and B cells in spinal cords was performed using an automated staining system (BOND-III; Leica Microsystems, Vista, CA) with antibodies against mouse CD4 (AbcamTM, dilution 1:200) and CD45R (eBioscience, dilution 1:300). Specificity of the primary antibodies was cross-validated. Omission of the primary antibodies was also used to test the specificity of the secondary antibodies. Isotype controls and secondary antibody only controls were included in each experiment. Digital images were acquired by Aperio CS2 using ImageScope viewing software, and the infiltration of T-cells and B-cells in spinal cord was quantified by counting positive cells using Aperio Image Analysis Systems.
Cytokine analysis
Whole blood of normal control and MOG1-128-induced EAE mice was collected by cardiac puncture and centrifuged at 1000g for five minutes to extract the serum at the endpoint of animal experiment. The serum levels of IgA and IL-17 were quantified by ELISA kits (Bio-techne, R&D systems) according to manufacturer’s instructions. In ELISA assay, serum was diluted by 1:200 and 1:1 for IgA and IL-17 detection, respectively. ODs at 450nm were measured by multimode plate reader (PerkinElmer EVision 2105).
Statistical analysis
Statistical analysis was conducted by GraphPad Prism 7.0 software. Data were expressed as mean ± standard error of mean (SEM). Normality and variance homogeneity of the data were assessed by shapiro-wilk test and kolmogorow smirnov test. Statistical comparisons of EAE clinical scores were conducted using two-way ANOVA, followed by Dunnett’s multiple comparisons test. A P-value of <0.05 was defined as statistically significant.
Results
T cell-modulating drugs and B cell-depleting agents exhibit distinct pharmacologic responses in MOG35-55 peptide-induced EAE model
MOG35-55 peptide-induced EAE models in wild-type C57BL/6 mice were utilized to evaluate the pharmacologic activities of BTKi-PRN2246 and S1P1 inhibitor Fingolimod. Clinical evaluation revealed that mice in vehicle control group of MOG35-55 peptide-induced EAE displayed typical clinical symptoms, such as severe tail paralysis and total limb weakness, while Fingolimod treatment significantly reduced the clinical severity of EAE. PRN2246 exhibited relatively weaker therapeutic effect on EAE mice compared with Fingolimod (Figure 1A). Consistently, histological analysis demonstrated inflammatory cell infiltration and demyelination in the spinal cord of EAE mice (Figure 1B & 1C). In addition, hIL17A/hIL17F mice were immunized with MOG35-55 peptide to evaluate the pharmacologic activities of Secukinumab and Telitacicept. Secukinumab was found to significantly ameliorate EAE clinical score, possibly through modulation of T cell function by neutralizing IL-17A. However, the clinical symptoms of EAE mouse treated with Telitacicept were significantly aggravated compared with vehicle control (Figure 1D).
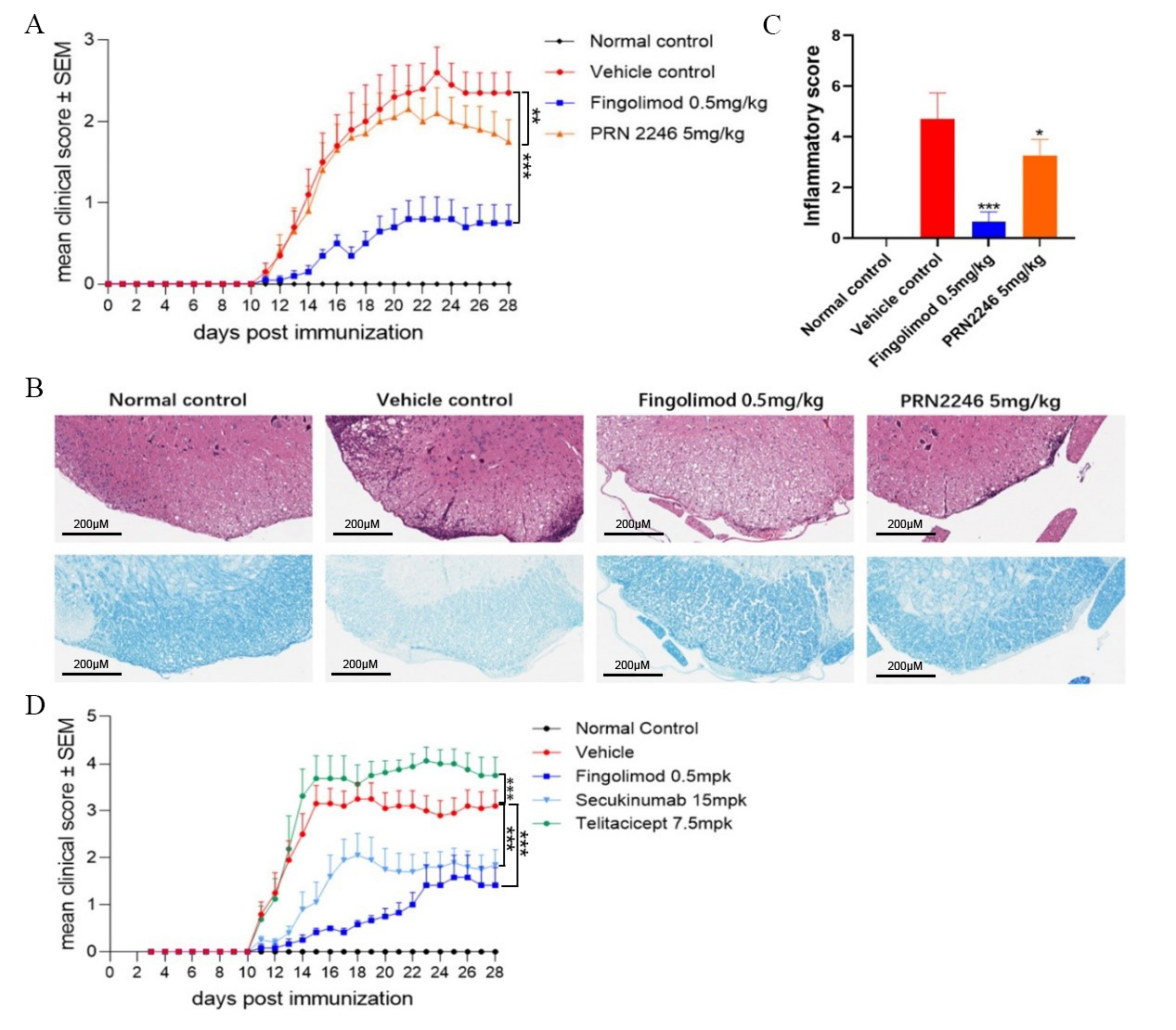
Figure 1: Pharmacologic responses and histological findings in MOG35-55-induced EAE mice treated with various anti-inflammatory agents.
(A) Clinical score of EAE mice receiving daily treatment with Fingolimod, PRN2246, or corresponding solvent. Asterisks indicate a significant difference between vehicle and Fingolimod or PRN2246 treatment groups. **p<0.01, ***p<0.005.
(B) Representative figures of H&E & fast blue staining of spinal cord in normal and EAE mice treated with Fingolimod, PRN2246, or corresponding solvent.
(C) Evaluation of inflammatory infiltration score in spinal cord by H&E staining. Graph represents the average score of five mice in each group. Asterisks indicate a significant difference between vehicle and Fingolimod or PRN2246 treatment groups. *p<0.05, ***p<0.005.
(D) Clinical score of EAE mice receiving daily treatment with Secukinumab, Telitacicept, or corresponding solvent. Graph represents the average score of 10 mice in each group. Asterisks indicate the significant difference between vehicle and Fingolimod, Secukinumab or Telitacicept treatment groups. ***p<0.005.
Collectively, all the results above demonstrated that drugs targeting T cells rather than B cells were therapeutically effective in MOG35-55-induced EAE model, suggesting that the pathogenesis of this model is dominated by T cells and it is unsuitable for evaluating pharmacological activities of B cell-depleting agents
Both B cell-depleting agents and T cell-modulating drugs show therapeutic response in MOG1-128-induced EAE model
In order to elucidate how drugs targeting different lymphocytes play a role during EAE induction by MOG1-128 protein, mice were independently treated with Evobrutinib and Fingolimod at the beginning of immunization process. As shown in Figure 2A, both Evobrutinib and Fingolimod had significant effects on relieving clinical symptoms of EAE. In addition, serum cytokine levels of EAE mice were also measured at the endpoint of the experiment. ELISA analysis revealed significantly increased IL-17 and IgA levels in vehicle control group compared to normal mice (Figure 2B), suggesting that MOG1-128 protein may trigger the upregulation of inflammatory factors, including IL-17 and IgA, which may be secreted by activated B cells and T cells.
Because innate and adaptive immune cells are thought to contribute to neuro-axonal injury and demyelination through the secretion of soluble factors during MS relapses [22], the pathogenesis of MOG1-128-induced EAE model may reflect real-life clinical situations in MS, while the EAE induction in humanized mice may be optimal for drug testing. To confirm this, we immunized IL-17A humanized mouse with MOG1-128 protein to induce EAE, and evaluated the pharmacological effects of Secukinumab and Telitacicept. Since human TACI protein is homologous in mice, the IL-17A humanized mouse is also suitable for testing the drug effect of Telitacicept on EAE model. Continuous observation of clinical symptoms revealed that both Secukinumab and Telitacicept reduced the severity of tail paralyses and limb weakness in mice, but Secukinumab showed higher efficacy than equal dose of Telitacicept (Figure 2C). Histological analysis also revealed that the number of infiltrating inflammatory cells around the white matter in spinal cord was significantly decreased after treatment with Secukinumab or Telitacicept (Figure 2D & 2E).
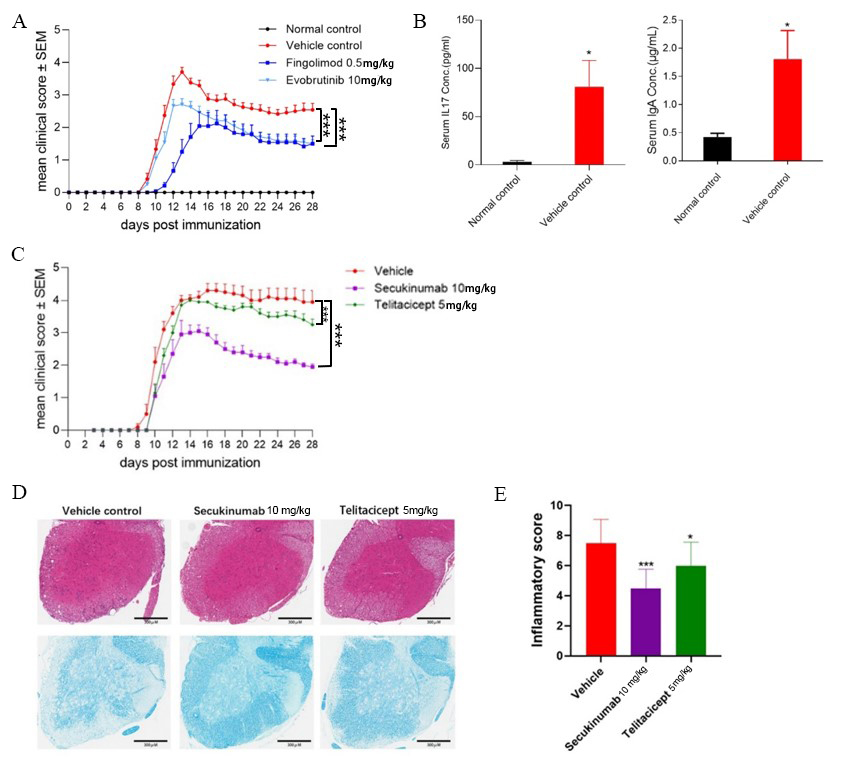
Figure 2: Pharmacologic responses, histological findings, and serum cytokine levels in MOG1-128 -induced EAE mice treated with various anti-inflammatory agents.
(A) Clinical score of EAE mice treated with Fingolimod, Evobrutinib, or corresponding solvent daily. Values are mean ± SEM of 10 mice/group. Asterisks indicate a significant difference between vehicle and Fingolimod or Evobrutinib treatment groups. ***p<0.005.
(B) Protein levels of IL-17 and IgA in serum of EAE mice. Values are mean ± SEM of five mice/group. *p<0.05 between vehicle control versus normal control.
(C) Clinical score of MOG1-128-induced EAE in hIL17A/hIL17F mice treated with Secukinumab, Telitacicept, or corresponding solvent daily. Values are mean ± SEM of 10 mice/group. Asterisks indicate a significant difference between vehicle and Secukinumab or Telitacicept treatment groups. ***p<0.005
(D) Representative figures of H&E staining of spinal cord in normal and EAE mice treated with Secukinumab, Telitacicept, or corresponding solvent.
(E) Evaluation of inflammatory infiltration score in spinal cord by H&E staining. Values are mean ± SEM of 10 mice/group. Asterisks indicate a significant difference between vehicle and dosing groups. *p<0.05, ***p<0.005.
Overall, the results suggest that molecules that suppress both B cell and T cell functions may have a considerable therapeutic effect on MOG protein-immunized mice, and the higher efficacy of Secukinumab and Telitacicept is suggestive of the vital function of Th17 cells and B cells in MOG1-128-induced EAE model, which may be used as an ideal model for screening and testing drugs for clinical treatment.
T cell and B cell infiltration in spinal cord increases with disease progression in MOG1-128 protein-induced EAE model
To further investigate the possible responding mechanism of T cells and B cells in MOG1-128 protein-induced EAE models, mice were sacrificed at different time points of the pathogenic process and the spinal cords were collected, sectioned, and stained. Hematoxylin & eosin (H&E) staining analysis revealed that only a few inflammatory cells had infiltrated into the gray matter of spinal cord at day 9 after immunization. However, the number of infiltrating cells had significantly increased 13 days post modeling. High-level cell infiltration remained constant until day 22 with the increased presence of inflammatory cells scattered in the central area of the gray matter (Figure 3A & 3B). We also calculated the total counts of T helper cells and B cells in the cervical, thoracic, and lumbar regions of spinal cord sections. As shown in Figure 3C, both CD4+ and CD45R+ cells started to accumulate since day 9, peaked at day 13, and remained constant until day 22 post immunization, which was similar to the H&E staining results. Collectively, the data show that T helper cell and B cell infiltration in the spinal cord increased with disease progression in MOG1-128 protein-induced EAE model, suggesting that both T and B cells contribute to the initiation and progression of this EAE model. Furthermore, T cell and B cell infiltration in the spinal cord from MOG35-55-induced EAE model mice was analyzed and we found that there is almost no B cell infiltration at day 9 (data not shown), suggesting B cells are not involved the pathogenic mechanism at the early stage of this model.
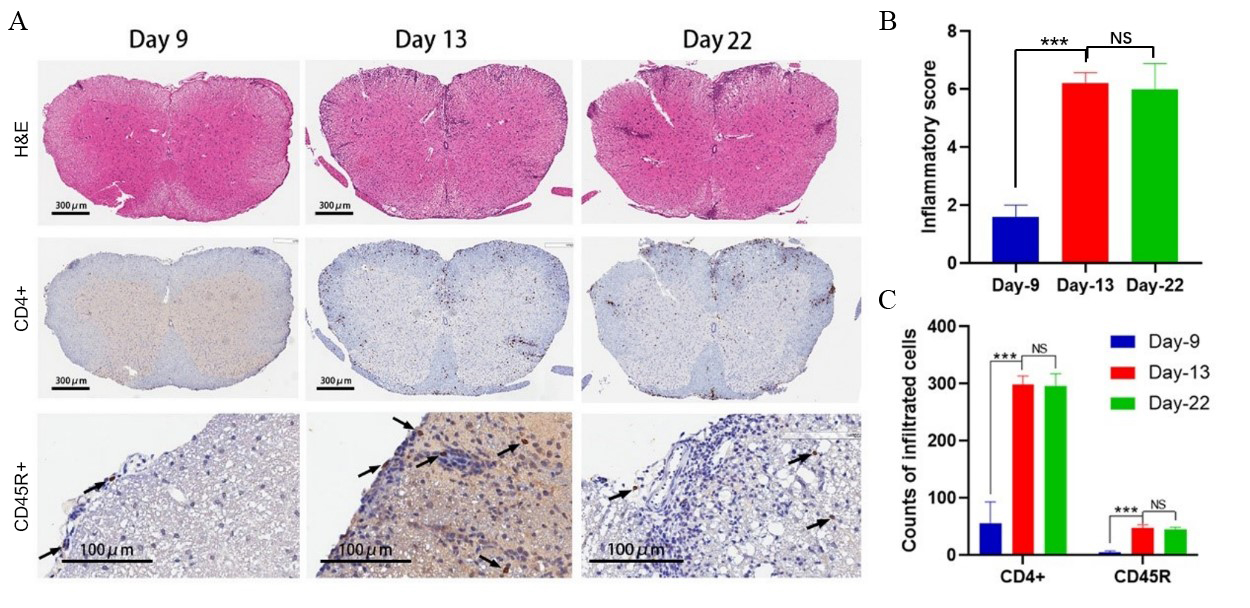
Figure 3: T helper cell and B cell infiltration analysis in the spinal cord of MOG1-128-induced EAE model.
(A) Representative figures of H&E and IHC staining at 9, 13, and 22 days after first immunization in the spinal cord of MOG1-128-induced EAE model.
(B) Evaluation of inflammatory infiltration score in spinal cord by H&E staining. Data shown on the graph represent the average score of five mice in each group. ***p<0.005
(C) Statistical IHC analysis of T helper cells (CD4+) and B cells (CD45R+) 9, 13, and 22 days after first immunization in the spinal cord of EAE mice. Values are mean ± SD of 5 mice/group. ***p<0.005.
Discussion
The immunological and neurobiological mechanisms underlying the pathogenesis, progression, and prognosis of MS are complicated. It is widely accepted that the hallmark of MS is the formation of demyelinating lesions in the brain and spinal cord, which are thought to be caused by infiltration of leukocytes including T cells, B cells, and myeloid cells into the lesion sites8, 16.
Animal models are important tools in research and pre-clinical drug development17. In this study, we established EAE models via induction with MOG p35-55 or MOG p1-128, and evaluated the pharmacological activities of a series of drugs for autoimmunity disease. These agents with distinct MOAs showed varied results in the two models due to differences in immunological mechanisms. Drugs modulating T cells exhibited therapeutic effects on both MOG35-55 peptide- and MOG1-128 protein-induced EAE models. However, agents targeting B cells only responded to MOG1-128-induced EAE model.
The MOG35-55-EAE C57BL/6 model is widely used due to the accessibility of short peptide and mature modeling methods. Consistent with previous study, our research demonstrated that Fingolimod, which sequesters T cells in lymph nodes, reduced EAE clinical score and inflammation of spinal cord. The serum level of the pro-inflammatory cytokine IL-17, mainly secreted by Th17 (a major subset of CD4+ T cell), was significantly increased in MS patients and EAE model18. In IL-17A humanized mouse, blocking the activity of IL-17A by Secukinumab alleviated clinical symptoms induced by MOG35-55. All these data suggested MOG35-55-induced EAE as an appropriate model for evaluating the pharmacological activities of agents targeting T cells activities. Nevertheless, this type of EAE model may not be a rationale choice when assessing more complex pathogenic immune responses involving both T and B cell recognition of autoantigen, since the antigen recognition of B cells depends on conformational epitopes of whole proteins.
The infiltration of B cells in active MS lesions19 and the success of B cell-depleting agents in MS therapy16, 20 confirm that B cells play a significant role in the pathogenesis of MS patients. In order to evaluate the therapeutic effects of agents targeting B cells, we immunized C57BL/6 mouse with the protein MOG1-128, which can be recognized by B cells. In this model, both B cell-depleting agents Telitacicept and Evobrutinib ameliorated clinical symptoms by suppressing B cell functions using different mechanism of actions. Notably, treatment with Telitacicept significantly aggravated MOG35-55-induced mouse EAE clinical symptoms, opposite to the effects in MOG1-128 model. The exacerbation of MOG35-55-induced EAE may relate to the depletion of antigen-naïve B cells, which exert anti-inflammatory properties21, 22. The paradoxical outcomes of Telitacicept in the two EAE models are similar to the results of a previous study on anti-CD20 agents23. BTK inhibitors, which may suppress both B cells and myeloid cells24, showed positive results in these two models, and the therapeutic effect in MOG35-55-induced EAE was mainly due to suppression of myeloid cells.
This study has three major limitations. First, the differences in immune responses to immunogenic MOG protein and peptide were not investigated in depth to clarify the complicated pathogenic mechanisms of these two EAE models. Second, the drugs/agents used in this study were limited and could therefore be biased. These agents may not completely represent the pharmacological mechanisms of therapeutic agents for MS treatment. Third, precautions should be applied when implying the conclusions obtained from this paper to addressing different aspects of questions in MS human patients. MS is a very complex disease and the pathogenesis is not completely known. The data from this paper provide preliminary guidance for animal model selection when addressing very specific mechanistic or pharmacological questions in preclinical studies, with the rodent–human dissimilarities and other potentially impactful factors born in mind. Therefore, more in-depth and systematic studies are needed to characterize the immunological mechanisms of these two paradigms of EAE models.
Conclusions
This study concludes that anti-inflammatory drugs with different MOAs exhibit differential pharmacological responses in EAE models induced by distinct immunogens. The MOG35-55 peptide-induced EAE model only responds to T cell modulating drugs, whereas the MOG1-128 protein-induced EAE model exhibits therapeutic sensitivity to both T cell- and B cell-modulating agents. These data indicate that it is critical to understand the pathogenesis of EAE animal models and provide preliminary rationale for model selection in MS research.
References
- Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med. 2018; 378: 169-180.
- Wei W, Ma D, Li L, et al. Progress in the Application of Drugs for the Treatment of Multiple Sclerosis. Front Pharmacol. 2021; 12: 724718.
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006; 1: 1810-1819.
- Lyons JA, San M, Happ MP, et al. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999; 29: 3432-3439.
- Dang AK, Jain RW, Craig HC, et al. B cell recognition of myelin oligodendrocyte glycoprotein autoantigen depends on immunization with protein rather than short peptide, while B cell invasion of the CNS in autoimmunity does not. J Neuroimmunol. 2015; 278: 73-84.
- von Budingen HC, Tanuma N, Villoslada P, et al. Immune responses against the myelin/oligodendrocyte glycoprotein in experimental autoimmune demyelination. J Clin Immunol. 2001; 21: 155-170.
- Constantinescu CS, Farooqi N, O'Brien K, et al. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011; 164: 1079-1106.
- Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015; 15: 545-558.
- Owens TD, Smith PF, Redfern A, et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin Transl Sci. 2021.
- Caldwell RD, Qiu H, Askew BC, et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton's Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J Med Chem. 2019; 62: 7643-7655.
- Ding J, Cai Y, Deng Y, et al. Telitacicept Following Plasma Exchange in the Treatment of Subjects With Recurrent NMOSD: Study Protocol for a Single-Center, Single-Arm, Open-Label Study. Front Neurol. 2021; 12: 596791.
- Frieder J, Kivelevitch D, Menter A. Secukinumab: a review of the anti-IL-17A biologic for the treatment of psoriasis. Ther Adv Chronic Dis. 2018; 9: 5-21.
- Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010; 9: 883-897.
- Tan YV, Abad C, Lopez R, et al. Pituitary adenylyl cyclase-activating polypeptide is an intrinsic regulator of Treg abundance and protects against experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2009; 106: 2012-2017.
- Seno A, Maruhashi T, Kaifu T, et al. Exacerbation of experimental autoimmune encephalomyelitis in mice deficient for DCIR, an inhibitory C-type lectin receptor. Exp Anim. 2015; 64: 109-119.
- Krumbholz M, Derfuss T, Hohlfeld R, et al. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. 2012; 8: 613-623.
- Denayer T, Stöhr T, Roy MV. Animal models in translational medicine: Validation and prediction. European Journal of Molecular & Clinical Medicine. 2014; 2.
- Babaloo Z, Aliparasti MR, Babaiea F, et al. The role of Th17 cells in patients with relapsing-remitting multiple sclerosis: interleukin-17A and interleukin-17F serum levels. Immunol Lett. 2015; 164: 76-80.
- Prineas JW, Connell F. The fine structure of chronically active multiple sclerosis plaques. Neurology. 1978; 28: 68-75.
- Chisari CG, Sgarlata E, Arena S, et al. Rituximab for the treatment of multiple sclerosis: a review. J Neurol. 2022; 269: 159-183.
- Evans JG, Chavez-Rueda KA, Eddaoudi A, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007; 178: 7868-7878.
- Fillatreau S, Sweenie CH, McGeachy MJ, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002; 3: 944-950.
- Weber MS, Prod'homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010; 68: 369-383.
- Burger JA. Bruton Tyrosine Kinase Inhibitors: Present and Future. Cancer J. 2019; 25: 386-393.