
HBsAg and Immune Competency; is HBsAg a Mere Biomarker or a Therapeutic Target for Chronic Hepatitis B?
Jin Hyang Kim1,2#, Grace Park3, Bhavna Paratala1,4, Rene Rijnbrand1,5, Michael J. Sofia1 and Hie-Won Hann3*
1Arbutus Biopharma Corporation, 701 Veterans Circle, Warminster, Pennsylvania, 18974, US
2Current address: Bristol Myers Squibb, 3401 Princeton Pike, Lawrenceville, NJ 08648, US
3Division of Gastroenterology and Hepatology, Department of Medicine, Thomas Jefferson University Hospital, Philadelphia, PA 19107, US
4Current address: Adaptimmune LLC, 351 Rouse Boulevard, Philadelphia, PA 19112, US
5Current address: Dicerna Pharmaceuticals, 33 Hayden Ave, Lexington, MA 02421
Abstract
Hepatitis B virus (HBV) is a main cause of hepatocellular carcinoma (HCC) development. Although controversial, it is increasingly recognized that the immune responses directed against infected hepatocytes drive hepatic inflammation and tissue injury. Here we extended our previous findings to report that serum surface antigen (HBsAg) levels are a biomarker not only for HBV-specific immunity, but also for ongoing non-specific immune activation. We found that the HBV-specific T cell responses in patients with HBsAg < 500 IU/mL, while significantly higher than those in patients with HBsAg > 50,000 IU/mL, had already reached levels comparable to patients with seroclearance. In addition, lower HBsAg levels were associated with reduced non-specific immune activation, while no further reduction was observed with HBsAg < 500 IU/mL. We propose HBsAg is a therapeutic target for reducing inflammation.
Introduction and Results
Chronic hepatitis B (CHB) infection is a major health burden due to the high morbidity and mortality. Presently, the World Health Organization estimates that 257 million people live with CHB and more than 850,000 deaths are attributed to CHB-mediated advanced liver diseases such as cirrhosis and HCC1. Current first line oral treatment is nucleot(s)ide analogues (NA), represented by tenofovir and entecavir which efficiently suppresses viral replication with low rates of drug resistance. Accordingly, a long-term entecavir treatment reduces HCC incidence2. However, the risk of HCC remains despite life-long treatment3-5, because NAs do not eliminate stably established covalently closed circular DNA (cccDNA), a template for persistent viral infection as well as viral antigen expression6. Additionally, integrated HBV DNA into the host genome further contributes to high viral antigenemia by consistently producing HBsAg7.
Chronic hepatic inflammation and ongoing tissue regeneration is a main driver for cirrhosis and HCC. While virus-driven events such as integration of HBV DNA into the hepatocyte genome and oncoproteins HBx and PreS/S mutants play a role for tumorigenesis8, it has been increasingly accepted that HBV-specific CD8 T cell responses and non-antigen-specific responses contribute to the necroinflammation and liver injury9, 10. In particular, recent studies demonstrated that blockade of a co-inhibitory receptor of T cells, T cell immunoglobulin and immune receptor tyrosine-based inhibitory motif domain (TIGIT), unleashed CD8 T cell responses and caused chronic inflammation and fibrosis in HBsAg-transgenic mice11.
The molecular entity that triggers chronic inflammation and immune activation remains elusive. Levels and profile of HBV-specific T cell responses are linked to the viral DNA load. Longitudinal studies showed that an HBV DNA load of < 107 copies/ml is a threshold at which envelope, core or polymerase-specific CD8 T cells responses are consistently detectable, whereas only core and envelope-specific T cells are occasionally found above this threshold12. HBV-specific T cell function is also suppressed at high DNA levels, with elevated expression of immune check-point proteins (CTLA4, PD1 and TIM3) as compared to their counterparts at the low viral DNA levels13. HBV DNA levels typically correlates with HBeAg and HBsAg in HBeAg(+) patients, but poorly in HBeAg(-) patients14. Thus, HBV DNA by itself may not be a sufficient guide for treatment15 or a biomarker for HBV-specific immune competency. In contrast, the levels of HBsAg are persistently high, with levels generally higher in HBeAg(+) patients than HBeAg(-) patients. Accordingly, we found that the HBsAg levels were stable over 10 years regardless of NA treatment or HBeAg status, with an approximate 1 log difference between HBeAg(+) and HBeAg(-) patients (Figure 1A). For patient with HBeAg seroconversion, HBeAg seropositivity fluctuated in the span of 5~10 years, while HBsAg levels were stable. Given the role of immune-mediated liver injury and stable HBsAg levels over the course of disease, we probed the possibility that HBsAg levels could be associated with specific and non-specific immune responses. HBsAg circulates in CHB patients as soluble proteins on fully infectious viral progeny, as well as constituents of non-infectious subviral particles (SVPs). Importantly, HBsAg is highly immunogenic, that patient-derived SVPs was successfully used in the first HBV vaccine and recombinant yeast-derived HBsAg has been subsequently a main component of the HBV vaccine16. HBsAg vaccine-induced immune memory, HBsAb responses and their association with clinical protection has been well established17, 18.
Given the potent immunogenicity of HBsAg, stable HBsAg-antigenemia of CHB patients appears at first enigmatic. Indeed, HBsAb has been detected in CHB patients19 and in our studies (unpublished). However, the biological role of HBsAg in the context of immune stimulation has been largely unappreciated. We have recently demonstrated that serum HBsAg levels were associated with HBV-specific immune competency20. In CHB patients with HBsAg levels > 50,000 IU/ml, HBV-specific T cell responses were significantly lower compared to patients with low HBsAg levels (≤500 IU/ml). Thus, it is conceivable to hypothesize that HBsAg seroclearance may further enhance HBV-specific immunity, compared to HBsAg ≤500 IU/ml. In this study, using rare cases of HBsAg-seroclearance patients, we found that HBV-specific T cell responses in patients with seroclearance were rather comparable to patients with HBsAg ≤500 IU/ml (p=0.14), though the proportion of responders was higher in patients with seroclearance (p=0.03) (Figure 1B). In addition, the responses to checkpoint blockade with αPD-L1 were comparable between two groups of patients. In our previous study, HBsAg-specific B cell responses were not affected by HBsAg levels20. In the current study, we confirmed that HBsAg-specific B cell frequency or HBsAb production were not different regardless of serum HBsAg levels (Figure 1C). Further, the levels of inhibitory receptors such as PD-L1 or FcRL5 were not only comparable between two groups but also within range of normal healthy donors (boxes, Figure 1C). Collectively, these observations suggest that HBV-specific immunity improves with the reduction of serum HBsAg levels and that serum HBsAg levels of 500 IU/ml appear to be a plateau point from which further recovery of HBsAg-specific immunity, if any, is negligible.
Figure 1:HBsAg levels were associated with HBV-specific immunity and non-specific immune activation.

A: Longitudinal serum HBsAg levels of CHB patients. Serum samples (n=18) were longitudinally archived from patients with HBeAg (+) (n=5), HBeAg-seroconversion (n=6) and HBeAg (-) (n=7) status. For each CHB patient, 4-6 samples were selected, each specimen 2-5 years apart, spanning over 10 -15 years. Year 0 indicates the first available serum specimen from each subject, which ranged 1-41 years after first diagnosis, not the baseline of treatment. The median treatment duration is 15.6 years. For patients with HBeAg seroconversion, HBeAg seropositivity fluctuated in the span of 5~10 years.
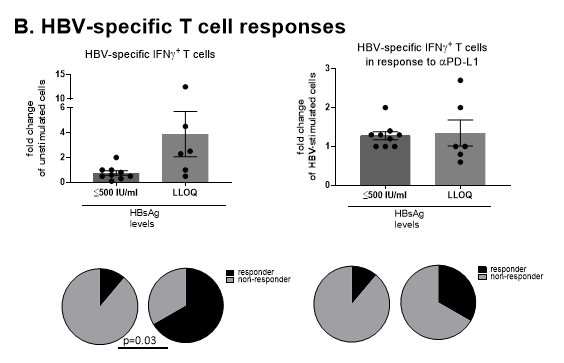
B: HBV-specific T cell responses of HBsAg-seroclearance. Peripheral blood mononuclear cells (PBMCs) from HBsAg-seroclearance patients (n=6) and patients with HBsAg ≤500 IU/ml (n=9) were stimulated with HBcAg and HBsAg in the absence (left) or presence of αPD-L1 antibody (right), followed by IFNγ-specific ELISpot assay. Fold changes of specific responses over unstimulated cells (left) or over HBV-stimulated cells (right) are shown. Responders were defined as patients with ≥ 2-fold change in IFNγ-secreting cells. Unpaired t test or χ2-test was performed for parametric data or association, respectively.
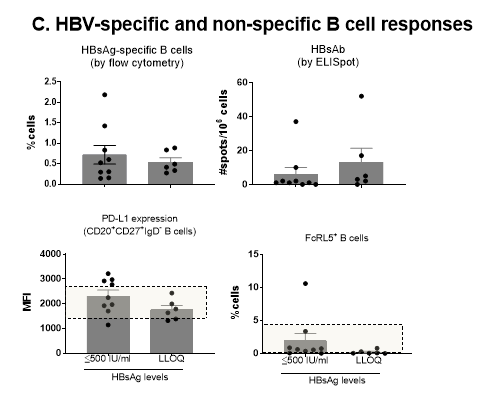
C: HBV-specific B cell responses of HBsAg-seroclearance. PBMCs from HBsAg-seroclearance patients (n=6) and patients with HBsAg ≤500 IU/ml (n=9) were analyzed for the HBsAg-specific B cell frequency, the HBsAb responses and the levels of cell surface PD-L1 and FcRL5 expression. Difference between groups was tested by unpaired t-test.
The apparent association between serum HBsAg levels and HBV-specific immunity leads to an important question as to whether HBsAg is merely a biomarker for HBV-specific immune competency or whether a HBsAg reduction is required to achieve seroclearance, a clinical milestone for functional cure. From the perspective of the immunogenicity of HBsAg, we asked whether HBsAg could affect the overall immune activation status in addition to HBsAg-specific immunity. We probed cellular as well as soluble markers that indicates overall non-specific immune activation (Figure 1D). Here, we included data obtained from CHB patients with a HBsAg >50,000 IU/ml for a better appreciation on their downward trend. We found that the CD69 expression on CD4 and CD8 T cells were significantly lower in HBsAg-seroclearance patients in comparison to patients with high HBsAg levels (>50,000 IU/ml), while there was a step-wise downtrend in its expression as HBsAg levels reduced. In addition, negative regulators of B cell activation including soluble CD40 (sCD40)21 and soluble FcgRIIB (sFcgRIIB)22 were significantly higher in patients with high HBsAg levels than in low HBsAg level (≤500 IU/ml) or seroclearance patients, indicating a significant global B cell activation is ongoing at high HBsAg levels. Further, several soluble co-stimulatory molecules critical for T and B cell interaction, including ICOS, CD28, CD86 and CD27 and NK cell frequencies were significantly higher in HBsAg > 50,000 IU/ml than HBsAg low or seroclearance, while comparable between the two latter groups (data not shown). Collectively, these findings suggest that significant global immune activation is ongoing at high HBsAg levels, but is restored at HBsAg ≤500 IU/ml at levels comparable to seroclearance. While this association does not directly prove a causal relationship, it adds plausibility that reducing HBsAg levels may bring additional benefit in reducing inflammatory milieu and restoring immune homeostasis.
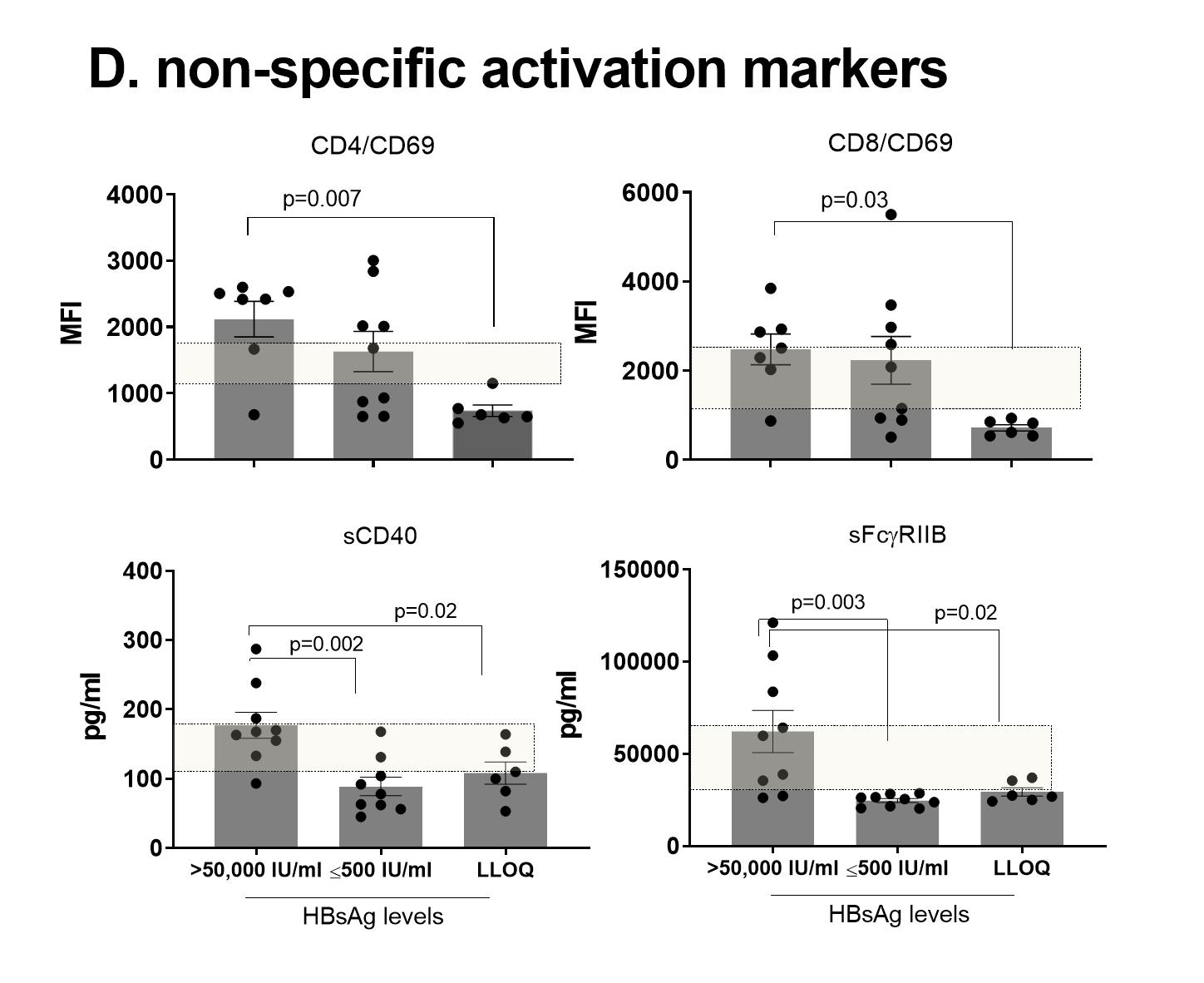
D: Cellular or soluble markers of immune activation. PBMCs from HBsAg-serocleared CHB patients (n=6) and patients with detectable HBsAg levels (>50,000 (n=7) and ≤500 IU/ml (n=9) were analyzed for cell surface CD69 expression on CD4 or CD8 T cells by flow cytometry. Soluble markers of serum samples were measured by LUMINEX or ELISA. Difference among groups was tested by one-way ANOVA with multiple comparison. The box indicates the levels of normal healthy volunteers (n=20). MFI; mean fluorescence intensity, LLOQ; lower limit of quantification.
Discussion
Based on our preliminary findings, we propose a model where serum HBsAg levels modulate HBV-specific and non-specific responses. At high HBsAg levels, HBV-specific T and B cells were chronically activated, yet counterbalanced ineffectively by elevated checkpoint receptor expression to protect tissues from immune-mediated injury. High HBsAg levels are also associated with global activation of non-HBV-specific T and B cells, hepatic NK cells or macrophages, further contributing to chronic hepatic inflammation. As HBsAg levels decrease, the fitness of HBV-specific responses increases with a reset of negative feedback mechanism, while the global activation is stabilized to reduce local inflammation. How HBsAg would trigger inflammation is not immediately clear. Interestingly, however, recent studies using HBsAg-transgenic mice demonstrated that HBsAg is a potent inflammatory trigger capable of inducing non-resolving chronic inflammation leading to HCC in the absence of immune checkpoint feedback mechanism11. In addition, non-specifically activated T cells through co-stimulation via CD137 (4-1BB) were sufficient to cause chronic fibrosis in HBsAg-transgenic mice23. Humoral immunity may also participate in the chronic hepatic inflammation. Both in our studies and other chronic viral infectious diseases, the B cell function and Ab production is not compromised20, 24. In the face of large amounts of HBsAg on the surface of HBV-infected hepatocytes 25, circulating HBsAb could induce CD16+ NK cell-mediated antibody-dependent cellular cytotoxicity, given the fact that HBsAb is capable of inducing a potent NK cell activation26 and that CD16+ NK cell frequencies are elevated in CHB patients27. Taken together, a pharmacological reduction of HBsAg may increase the functional cure rate and reduce hepatic inflammation and long-term tissue deterioration of CHB patients.
Limitation
We acknowledge that the sample size is small in the current study, therefore the findings are not likely conclusive and warrants further studies. Spontaneous or treatment-induced HBsAg seroclearance is a rare clinical event, and collecting longitudinal peripheral blood mononuclear cells (PBMCs) presents a significant difficulty in clinical practice due to long duration of the disease and stability of HBsAg levels. Additional limitations include single center patient cohort and retrospective design, which may have affected the patient selection. Some samples were obtained from commercial sources for comparison, but unavailability of full laboratory safety assessment including alanine aminotransaminase (ALT) or histology limited further exploratory correlation analyses. From the analytical perspective, large scale orthogonal approaches including genomics and proteomics may be needed to elucidate the cellular and soluble factors’ contribution to inflammation and tissue damage. In this short communication, building from collective evidence from our recent report20 and the current preliminary findings, we propose a hypothesis that HBsAg represents not only a biomarker whose levels ≤500IU/ml appears to be a threshold for restoration of HBV-immunity competency, but also a potential inflammatory trigger for which a pharmacological reduction may confer better clinical prognosis to CHB patients. With HBsAg-reducing therapeutic agents in clinical development, such proof of concept may become realized in the near future.
Materials and Methods
Patients: CHB patients under routine care of one author at the Thomas Jefferson University were included in this study, with average disease duration over a decade for most patients. Patients with HCC or decompensated cirrhosis or coinfected with HIV, HCV or HDV were excluded. All subjects were Asian Americans with the HBV genotype C for most patients. Depending on the time of first diagnosis, the antivirals consisted of lamivudine, adefovir, entecavir, telbivudine, tenofovir disoproxil fumarate or tenofovir alafenamide. Archived serum samples from a total of 18 CHB patients (Figure 1), who were on antiviral therapy for a median 15.6 years, were used to assess the serum HBsAg levels. No specific laboratory abnormality including albumin, ALT, white blood cell (WBC) counts, platelet counts, or alpha-fetoprotein was noted from the patients, except 1 HBeAg (-) patient whose average platelet counts were low (95K) but stable. Abdominal ultrasounds obtained during follow-up for all patients showed no liver tumor. The study protocol was approved by the Institutional Review Board at the Thomas Jefferson University and all subjects have provided written informed consent. Additional CHB samples were obtained from Sanguine Bioscience Inc. (Los Angeles, CA) or bioIVT (Westbury, NY) for comparison.
HBsAg quantification, antibody staining, flow cytometry and the assessment of the HBV-specific T cell and B cell response were previously described20. Briefly, HBV-specific T cell responses were measured by IFNγ-specific ELISpot assay following stimulation of PBMCs with overlapping peptides of HBcAg and HBsAg (1mg/ml each) for 24hr. Responses to check-point blockade was assessed in the absence or presence of αPD-L1 antibody during peptide stimulation. HBsAg-specific B cell frequency was measured by flow cytometry with fluorescence-labeled rHBsAg. HBsAg-specific Ab responses were measured by HBsAb-ELISpot following stimulation of PBMCs with CpG-B for 5 days. The expression of inhibitory receptors (PD-L1 or FcRL5) on B cells was measured by flow cytometry.
Soluble markers were measured using Milliplex Map Human Immuno-Oncology Checkpoint protein panel (Millipore Sigma) as previously described20. Additional soluble marker including sFcgRIIB were measured by ELISA according to manufacturer’s instruction (ThermoFisher).
Conflict of Interest: MS is employed by Arbutus Biopharma Corporation. JK, BP and RR were employed by Arbutus Biopharma at the time of manuscript preparation, are currently employed by Bristol Myers Squibb, Adaptimmune and Dicerna Pharmaceuticals, respectively. HWH serves at the National Advisory Board of Gilead, received research grant from Gilead, Assembly Biosciences and Arbutus. GP declared there is no conflict of interest.
Acknowledgement: We thank the patients who provided clinical samples.
Ethics statement: All participants signed informed consent approved by Thomas Jefferson University at the time of sample collection. All samples were anonymized.
References
- World Health Organization, G.H., Hepatitis and Sexually Transmitted Infections Programms, Global hepatitis report. 2017.
- Papatheodoridis, G.V., et al., Incidence of hepatocellular carcinoma in chronic hepatitis B patients receiving nucleos(t)ide therapy: a systematic review. J Hepatol, 2010. 53(2): p. 348-56.
- Kim SU, et al., Hepatocellular Carcinoma Risk Steadily Persists over Time Despite Long-Term Antiviral Therapy for Hepatitis B: A Multicenter Study. Cancer Epidemiol Biomarkers Prev, 2020. 29(4): p. 832-837.
- Russo FP, et al., Hepatocellular Carcinoma in Chronic Viral Hepatitis: Where Do We Stand? International Journal of Molecular Sciences, 2022. 23(1): p. 500.
- Shinn BJ, et al., Persistent risk for new, subsequent new and recurrent hepatocellular carcinoma despite successful anti-hepatitis B virus therapy and tumor ablation: The need for hepatitis B virus cure. World J Hepatol, 2019. 11(1): p. 65-73.
- Dandri M and J. Petersen, cccDNA Maintenance in Chronic Hepatitis B - Targeting the Matrix of Viral Replication. Infection and drug resistance, 2020. 13: p. 3873-3886.
- Pollicino T and G. Caminiti, HBV-Integration Studies in the Clinic: Role in the Natural History of Infection. Viruses, 2021. 13(3).
- Li Y-W., et al., Hepatocellular carcinoma and hepatitis B surface protein. World journal of gastroenterology, 2016. 22(6): p. 1943-1952.
- Rehermann B, Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med, 2013. 19(7): p. 859-68.
- Nakamoto Y., et al., Immune pathogenesis of hepatocellular carcinoma. The Journal of experimental medicine, 1998. 188(2): p. 341-350.
- Zong L, et al., Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nature Communications, 2019. 10(1): p. 221.
- Webster GJ., et al., Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: implications for immunotherapy. J Virol, 2004. 78(11): p. 5707-19.
- Boni C, et al., Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol, 2007. 81(8): p. 4215-25.
- Yang N., et al., Relationship between serum quantitative HBsAg and HBV DNA levels in chronic hepatitis B patients. J Med Virol, 2018. 90(7): p. 1240-1245.
- Bárcena Marugán R and S. García Garzón, DNA-guided hepatitis B treatment, viral load is essential, but not sufficient. World journal of gastroenterology, 2009. 15(4): p. 423-430.
- CDC, Recommendation of the Immunization Practices Advisory Committee (ACIP) Inactivated Hepatitis B Virus Vaccine. MMWR 1982. 31(24): p. 317-322, 327-328.
- Schillie S, V.C., Reingold A, et al., Prevention of Hepatitis B Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm Rep, 2018. 67(No. RR-1): p. 1-31.
- Wang R-X, et al., Long-term persistence of T cell memory to HBsAg after hepatitis B vaccination. World journal of gastroenterology, 2004. 10(2): p. 260-263.
- Gerlich WH, The Enigma of Concurrent Hepatitis B Surface Antigen (HBsAg) and Antibodies to HBsAg. Clinical Infectious Diseases, 2007. 44(9): p. 1170-1172.
- Kim JH, et al., Circulating serum HBsAg level is a biomarker for HBV-specific T and B cell responses in chronic hepatitis B patients. Scientific Reports, 2020. 10(1): p. 1835.
- Contin C, et al., Membrane-anchored CD40 is processed by the tumor necrosis factor-alpha-converting enzyme. Implications for CD40 signaling. J Biol Chem, 2003. 278(35): p. 32801-9.
- Esposito-Farese M.E, et al., Membrane and soluble Fc gamma RII/III modulate the antigen-presenting capacity of murine dendritic epidermal Langerhans cells for IgG-complexed antigens. J Immunol, 1995. 155(4): p. 1725-36.
- Wang J, et al., CD137-Mediated Pathogenesis from Chronic Hepatitis to Hepatocellular Carcinoma in Hepatitis B Virus-Transgenic Mice. The Journal of Immunology, 2010. 185(12): p. 7654-7662.
- Fallet B, et al., Chronic Viral Infection Promotes Efficient Germinal Center B Cell Responses. Cell Reports, 2020. 30(4): p. 1013-1026.e7.
- Yim SY, et al., Expression of Hepatocyte Hepatitis B Core Antigen and Hepatitis B Surface Antigen as a Marker in the Management of Chronic Hepatitis B Patients. Gut and liver, 2017. 11(3): p. 417-425.
- Li D, et al., A potent human neutralizing antibody Fc-dependently reduces established HBV infections. eLife, 2017. 6: p. e26738.
- Wijaya RS., et al., Expansion of dysfunctional CD56-CD16+ NK cells in chronic hepatitis B patients. Liver International. n/a(n/a).