
Identification of ω-sites of Glycosylphosphatidylinositol Anchored Proteins
Yusuke Masuishi*, Shota Endo, Hideaki Kasuga, Tomoo Hidaka, Takeyasu Kakamu, Tetsuhito Fukushima
Department of Hygiene and Preventive Medicine, Fukushima Medical University School of Medicine, 1Hikariga-oka, Fukushima City 960-1295, Japan
Abstract
Unique and complex post-translational modifications are present in the outer leaflet of the plasma membrane. Glycosylphosphatidylinositol (GPI) anchoring is essential for the expression of several outer membrane proteins on the cell surface. A common GPI anchor structure is constituted by glycan moiety, lipid moiety, phosphate and ethanolamine. GPI-anchored proteins (GPI-APs) are observed among eukaryotic species. Abnormal GPI anchoring of proteins is thought to cause various diseases such as paroxysmal nocturnal hemoglobinuria. Recently, many inherited GPI deficiencies (IGDs) have been reported to cause epilepsy, mental retardation, coarse facial features, and multiple organ anomalies. Diseases caused by abnormal GPI anchoring will probably continue to increase, because it is still unknown how many causative genes of IGDs are present. Therefore, in order to study these diseases, the analytical methods of GPI-APs will become important in the future. To date, many methods have been developed for analysis of GPI- APs. In this review, we attempt to summarize the present knowledge about comprehensive analytical methods of GPI-APs and introduce briefly some GPI anchor-related diseases.
Several outer membrane proteins contain a covalently linked glycosylphosphatidylinositol anchor (GPI-anchor), which tethers the protein to the extracellular face of the plasma membrane. The common GPI core structure is ethanolamine-phosphate Manα1−2Manα1−6Manα1−4GlcNα1−6myo-ino-1-phosphate-lipid, and is highly conserved among eukaryotic species. All mammalian GPI anchors have a phosphoethanolamine side chain linked to the 2-position of the first α1-4 linked mannose1 (Figure 1). Precursors of GPI-APs contain a C- terminal GPI-attachment signal peptide that is involved in the addition of GPI anchor by the transamidase complex in the endoplasmic reticulum2. GPI-anchor attachment site — the ω-site, tends to contain amino acids with small side chains, such as Gly, Ala, Ser, Asn, Asp, and Cys3. However, we revealed that other amino acids (Leu, Met and Thr) are also present at ω- sites4. GPI-APs are a diverse class of proteins that play critical roles in many biological processes such as immunity, inflammatory response, growth arrest, cell adhesion, lipid metabolism, fertilization, transport, and neurogenesis.
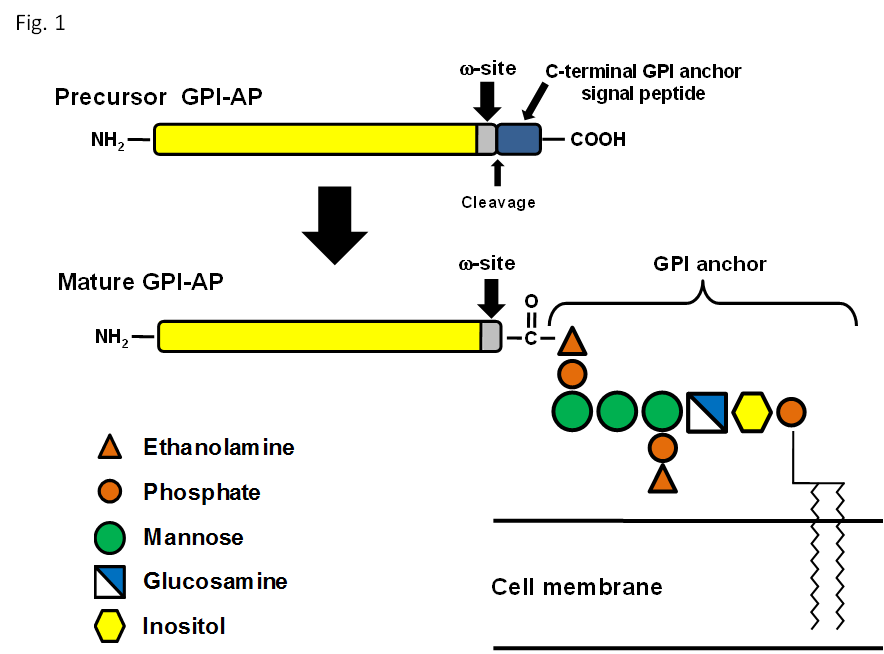
Figure 1: General schematic representation of precursor GPI-AP and mature GPI-AP.
The identification of phospholipid anchored alkaline phosphatase (ALP) was first reported by Slein, M. W. & Logan, G. F. in 1963 by using phospholipase C (PLC) in mammalian cells5. In human cells, four types of GPI-anchored ALP have been identified to date (ALPI, ALPL, ALPP, ALPPL2). In 1988, the first complete GPI-anchor structure was obtained for Trypanosoma brucei variant surface glycoprotein (VSG) and rat Thy-1.
Trypanosoma brucei evades its host’s adaptive immune response by VSG coat replacement6. To date, more than 150 proteins have been reported as a GPI-APs in mammals7, which play important roles in many biological processes. It is predicted that there might be 460 GPI-APs by genome analysis8.
GPI Anchor Related Diseases
The most famous GPI anchor related disease is paroxysmal nocturnal hemoglobinuria (PNH), which is a clonal hematopoietic stem cell disorder resulting from acquired somatic mutations in the PIG-A gene9. The PIG gene family encodes over 20 different proteins that are essential for the synthesis of GPI anchors. GPI-APs such as CD55 and CD59 are important complement regulatory proteins. In PNH patients, improper expression of CD55 and CD59 on blood cells leads to chronic complement-mediated intravascular hemolysis10. Eculizumab (Soliris) is a humanized monoclonal antibody that specifically binds to the complement protein C5. Eculizumab inhibits terminal complement activation and intravascular hemolysis, improving quality of life of PNH patients11.
The prion protein is a GPI-AP, ubiquitously expressed throughout the body. Creutzfeldt–Jakob disease (CJD) is the most common human prion disease and a fatal neurodegenerative disorder with hallmark features of dementia, ataxia, and myoclonus12. Prion diseases are neurodegenerative disorders characterized by changing its conformation from a cellular prion protein (PrPC) to a scrapie isoform of the prion protein (PrPSc)13-15. It was demonstrated that GPI-anchor is not required for PrPSc formation by using transgenic mice expressing a prion protein lacking the GPI-anchor16. However, in these PrPSc-infected mice, it developed a new type of prion disease with unique clinical signs and altered neuropathology17. Campana et al.18 reported that GPI anchorless prion protein is secreted in a soluble form, and does not acquire scrapie-like characteristics in transfected Fischer rat thyroid cells. Nisbet et al.19 reported that peptides of rabbit prion proteins surrounding the ω-site prevented prion infection. Gianni et al.20 reported that the C-terminal GPI-attachment signal peptide of PrPC might be responsible for a genetic form of prion diseases. Sarnataro et al. reported that lipid rafts components might be involved in the prion disease process21. Although, the roles of GPI anchor in PrPSc propagation and toxicity are unclear, GPI anchor might be involved in prion disease pathogenesis.
There are at least 26 genes involved in the biosynthesis of GPI-anchors and GPI-anchoring such as PIG genes in the endoplasmic reticulum of mammalian cells. Recently, many inherited GPI deficiencies (IGDs) have been found using whole-exome sequencing22-26. Mutations of these genes (PIGA, PIGL, PIGM, PIGN, PIGO, PIGP, PIGQ PIGT, PIGV, PIGW, PIGY, PIGAP1, PIGAP2 and PIGAP3) are known to cause IGDs, and it is likely that novel IGDs will be identified in the future. Several studies summarized individual clinical symptoms of IGDs27-29. IGDs show reduced surface levels of GPI-APs or abnormal GPI-AP structure and can be diagnosed by serum alkaline phosphatase (ALP) levels and flow cytometry of granulocytes30. IGDs are a group of rare diseases and the major symptoms of patients with IGDs include mental retardation and epilepsy. However, in some reported cases of IGDs, GPI-APs were expressed and localized normally31. This result indicates that the detail of IGDs is not yet fully understood.
Analytical Methods of GPI-APs
The photo imaging can be used to observe the lateral mobility of GPI-Aps on the cell surface. It is considered that the GPI anchor provides a high lateral mobility for GPI-APs by preventing interactions between the lipid bilayer and the protein moiety of GPI-APs. A useful fluorescence reagent for a GPI-AP analysis is fluorescent-labeled inactive toxin aerolysin (FLAER) that is tagged with Alexa Fluor 488 dye and binds the glycan moiety of the GPI anchor32. FLAER staining and FLAER based flowcytometry are used to define GPI-APs on the cell surface. Combining FLAER based flow cytometry with other multiparametric flow cytometry can further improve the diagnosis of PNH and other IGDs. However, in the erythrocyte lineage, FLAER cannot be used to diagnose PNH, since erythrocytes do not possess proteolytic enzymes needed to process the Proaerolysin33.
Because the GPI anchor is a complex structure, the identification of GPI- anchored sites by mass spectrometry has always been considered difficult. Recently, we developed a novel method for the identification of GPI- anchored sites by combining titanium dioxide (TiO2)–based affinity purification and hydrogen fluoride (HF) treatment. Figure 2 shows a schematic overview of strategies for identifying ω-site (GPI-anchored sites). Using this novel technique we conducted for the first time a comprehensive analysis of GPI-APs4. Moreover, by applying this technique, the glycan moiety of its GPI anchor can be analyzed. Using metabolic tagging, another approach, other researchers identified the GPI-APs of Plasmodium34.
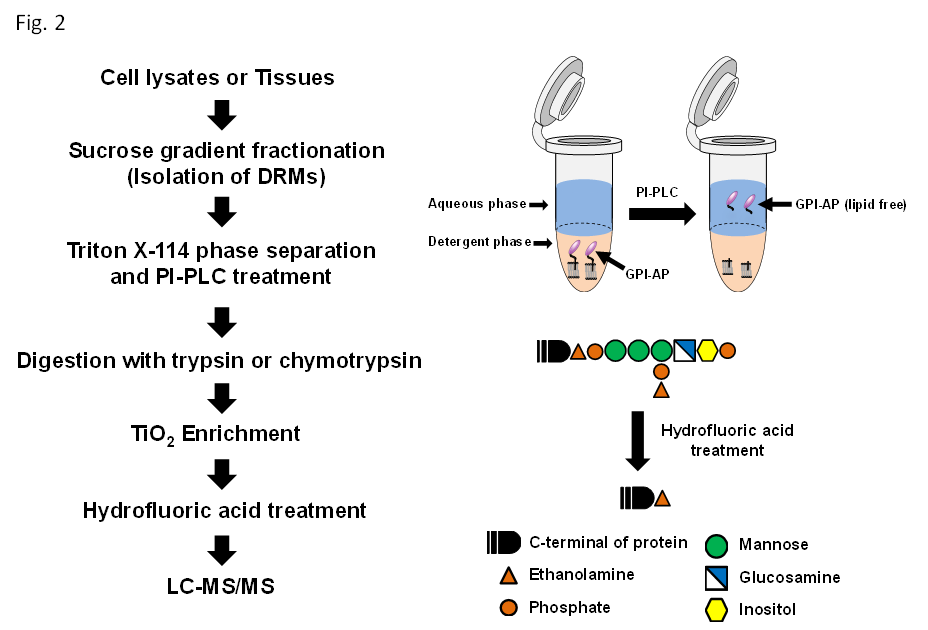
Figure 2: Schematic overview of the identification of GPI-APs, and schematic representation of Triton X-114 phase separation and hydrogen fluoride treatment.
Lipid rafts are micro domains enriched in GPI-APs cholesterol, sphingolipids, transmembrane proteins, and lipidated proteins35,36. There have been several reports that lipid raft associated proteins including GPI- APs are insoluble at 4 °C in nonionic detergents, such as Triton X-100, CHAPS, and Brij 96, and able to purify these proteins by using sucrose gradient centrifugation37,38. The phospholipid moiety of the GPI anchor is critical for the hydrophobicity of GPI-APs. The GPI anchor is cleaved between the phosphate moiety and the lipid moiety by treatment with a phosphatidylinositol-specific phospholipase C (PI-PLC) or phospholipase D (PLD). When subjected to temperature-induced phase separation in Triton X-114, the PI-PLC or PLD-cleaved hydrophilic forms of GPI-APs are found in the aqueous phase8. Elortza et al. isolated GPI-APs from Arabidopsis thaliana and human lipid raft associated proteins by PI-PLC and PLD treatment. This approach can indicate the presence of GPI-APs and identify ω-sites using mass spectrometric analysis. However, due to the complex structure of the GPI-anchor, comprehensive identification of the ω-sites was difficult on a proteomic scale.
The GPI anchor of VSG Trypanosoma brucei was isolated following aqueous HF treatment and nitrous acid deamination of the purified protein. HF treatment cleaves the phosphodiester bond of the GPI anchor moiety. Therefore, GPI anchor moiety is divided into protein moiety, glycan moiety and lipid moiety. The HF-treated GPI-APs contain only the ethanolamine of the GPI anchor (+43.0422 Da modification) (Figure 2). It is crucial to provide direct evidence for the identification of GPI-APs. In our previous study, we demonstrated that liquid chromatography–tandem mass spectrometry (LC- MS/MS) and protein database search can identify the HF-treated GPI- anchored peptides39, and our study is the first comprehensive enrichment analysis of GPI-APs. Recently, TiO2 is used to isolate phosphorylated peptides from digested peptides in phosphoproteomic analysis40. In comparison with IMAC purification, TiO2 is more selective for binding phosphate moiety of peptides41. In our previous study, we developed a method that uses PI-PLC treatment, HF treatment, TiO2-based affinity purification, and mass spectrometry. Using this method, we identified a total of 73 ω-sites derived from 49 GPI-APs. Interestingly, we found that 13 of the 49 identified GPI-APs had different amino acids at the ω-sites, yielding different protein species3. Several studies demonstrated that alteration of ω-site amino acids has a strong influence on the localization of GPI-APs. Therefore, normally, a function of GPI-APs almost certainly depends on the GPI anchor moiety in lipid rafts. The method for the identification of ω-sites could be used in a detailed analysis of GPI anchor related diseases.
References
- Orlean P, Menon AK. Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res. 2007; 48: 993–1011.
- Ferguson MA, Brimacombe JS, Brown JR, et al. The GPI biosynthetic pathway as a therapeutic target for African sleeping sickness. Biochim. Biophys Acta. 1999; 1455: 327-40.
- Gerber LD, Kodukula K, Udenfriend S. Phosphatidylinositol glycan (PI-G) anchored membrane proteins. Amino acid requirements adjacent to the site of cleavage and PI-G attachment in the COOH-terminal signal peptide. J Biol Chem. 1992; 267(17): 12168-73.
- Masuishi Y, Kimura Y, Arakawa N, et al. Identification of glycosylphosphatidylinositol-anchored proteins and ω-sites using TiO2- based affinity purification followed by hydrogen fluoride treatment. J Proteomics. 2016; 139: 77-83.
- Moser JB, Marshall GW, Green FP. Direct bonding of polycarbonate orthodontic brackets: an in vitro study. Am J Orthod. 1979; 75(1): 78-85.
- Cross GA, Kim HS, Wickstead B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol Biochem Parasitol. 2014; 195(1): 59-73.
- Fujita M, Kinoshita T. Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett. 2010; 584(9): 1670-7.
- Elortza F, Mohammed S, Bunkenborg J, et al. Modification-specific proteomics of plasma membrane proteins: identification and characterization of glycosylphosphatidylinositol-anchored proteins released upon phospholipase D treatment. J Proteome Res. 2006; 5: 935- 43.
- Kinoshita T, Inoue N, Takeda J. Role of phosphatidylinositol-linked proteins in paroxysmal nocturnal hemoglobinuria pathogenesis. Annu Rev Med. 1996; 47: 1-10.
- Parker CJ. Hemolysis in PNH. In: Young NS, Moss J, eds. Paroxysmal Nocturnal Hemoglobinuria and the Glycosylphosphatidylinositol-Linked Proteins. Academic Press. 2000; 49-100.
- Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004; 350(6): 552-9.
- Sitammagari KK, Masood W. Creutzfeldt Jakob Disease. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2018.
- Sandberg MK, Al-Doujaily H, Sharps B, et al. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011; 24470(7335): 540-2.
- Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005; 15(2): 102-11.
- Sarnataro D, Pepe A, Zurzolo C. Cell Biology of Prion Protein. Prog Mol Biol Transl Sci. 2017; 150: 57-82.
- Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005; 3308(5727): 1435-9.
- Chesebro B, Race B, Meade-White K, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010; 6(3): e1000800.
- Campana V, Caputo A, Sarnataro D, et al. Characterization of the properties and trafficking of an anchorless form of the prion protein. J Biol Chem. 2007; 3282(31): 22747-56.
- Nisbet RM, Harrison CF, Lawson VA, et al. Residues surrounding the glycosylphosphatidylinositol anchor attachment site of PrP modulate prion infection: insight from the resistance of rabbits to prion disease. J Virol. 2010; 84(13): 6678-86.
- Guizzunti G, Zurzolo C. The fate of PrP GPI-anchor signal peptide is modulated by P238S pathogenic mutation. Traffic. 2014; 15(1): 78-93.
- Sarnataro D, Campana V, Paladino S, et al. PrP(C) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol Biol Cell. 2004; 15(9): 4031-42.
- Pagnamenta AT, Murakami Y, Taylor JM, et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. 2017; 25(6): 669-679.
- Brady PD, Moerman P, De Catte L, et al. Exome sequencing identifies a recessive PIGN splice site mutation as a cause of syndromic congenital diaphragmatic hernia. Eur J Med Genet. 2014; 57(9): 487-93.
- Krawitz PM, Schweiger MR, Rödelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010; 42(10): 827-9.
- Johnstone DL, Nguyen TT, Murakami Y, et al. Compound heterozygous mutations in the gene PIGP are associated with early infantile epileptic encephalopathy. Hum Mol Genet. 2017; 126(9): 1706- 1715.
- Zhao JJ, Halvardson J, Knaus A, et al. Reduced cell surface levels of GPI-linked markers in a new case with PIGG loss of function. Hum Mutat. 2017; 38(10): 1394-1401.
- Bellai-Dussault K, Nguyen TTM, Baratang NV, et al. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin Genet. 2018; 27. [Epub ahead of print]
- Knaus A, Pantel JT, Pendziwiat M, et al. Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med. 2018; 910(1): 3.
- Ng BG, Freeze HH. Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J Inherit Metab Dis. 2015; 38(1): 171-8.
- Kuki I, Takahashi Y, Okazaki S, et al. Vitamin B6-responsive epilepsy due to inherited GPI deficiency. Neurology. 2013; 81(16): 1467-9.
- Makrythanasis P, Kato M, Zaki MS, et al. Pathogenic Variants in PIGG Cause Intellectual Disability with Seizures and Hypotonia. Am J Hum Genet. 2016; 98(4): 615-26.
- Brodsky RA, Mukhina GL, Li S, et al. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000; 114(3): 459-66.
- Sutherland DR, Kuek N, Davidson J, et al. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom. 2007; 72(3): 167-77.
- Wright MH, Clough B, Rackham MD, et al. Validation of N- myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat Chem. 2014; 6(2): 112-21.
- Milhiet PE, Giocondi MC, Baghdadi O, et al. Spontaneous insertion and partitioning of alkaline phosphatase into model lipid rafts. EMBO Rep. 2002; 3(5): 485-90.
- Schuck S, Simons K. Polarized sorting in epithelial cells: raft clustering and the biogenesis of the apical membrane. J Cell Sci. 2004; 117(25): 5955- 64.
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid- enriched membrane subdomains during transport to the apical cell surface. Cell. 1992; 68(3): 533-44.
- Parkin ET, Turner AJ, Hooper NM. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem J. 1999; 344(1): 23-30.
- Masuishi Y, Nomura A, Okayama A, et al. Mass spectrometric identification of glycosylphosphatidylinositol-anchored peptides. J Proteome Res. 2013; 12(10): 4617-26.
- Kuroda I, Shintani Y, Motokawa M, et al. Phosphopeptide-selective column-switching RP-HPLC with a titania precolumn. Anal Sci. 2004; 20(9): 1313-9.
- Larsen MR, Thingholm TE, Jensen ON, et al. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics. 2005; 4(7): 873-86.