
Immunometabolic Links Underlying the Infectobesity with Persistent Viral Infections
Yongming Sang*
Department of Agricultural and Environmental Sciences, College of Agriculture, Tennessee State University, 3500 John A. Merritt Boulevard, Nashville, TN, USA
Abstract
Obesity and its related comorbidities are prevailing globally. Multiple factors are etiological to cause obesity and relevant metabolic disorders. In this regard, some pathogenic infections including those by viruses have also been associated with obesity (termed as infectobesity). In this mini-review, I examined recent publications about primary or cofactorial role of viral infections to exacerbate the local and systemic immunometabolic cues that underlie most cofactorial obesity. Major immuno-metabolic pathways involved, including that mediated by interferon (IFN) signaling and peroxisome proliferator activated receptor-γ (PPAR-γ), are discussed.
Introduction
Obesity manifests as metabolic overload of excess fat in adipose depots, but entails various immunological disorders. This can be further worsen the overweight into a metabolic syndrome as well as other life-threatening complications including diabetes, heart disease, liver steatosis, and cancer1,2. Obesity and its related comorbidities are epidemic globally to reach an alarming level. Various factors from predisposing genetics to habitat environment could contribute to the complex disorders relevant to obesity epidemic1-3. Despite intensive studies, the etiological keys that cause metabolic disorders and obesity remain incompletely understood3. From an evolutionary view, animal metabolic and immune systems integrate as the most fundamental requirement for a species survival4. In this mini-review, I will focus on the obesity induced by energy-dense diets and viral agents to examine the major immunological links that drive relevant metabolic disorders and obesity development5.
Viral Infectobesity
Infectobesity describes the association of microbial infections with obesity and its relevant morbidities, which has been often observed in humans and reproduced in animal models5-8. The major viruses and virus-like agents (VLA) that have been associated with obesity include members in adenoviridae, herpesviridae, phages, transmissible spongiform encephalopathies (slow virus), and hepatitides5-8. For example, at least five human adenovirus (Ad) serotypes, i.e. Ad-5, -9, -31, -36, and -37, have been associated with obesity at different levels. These adenoviruses demonstrated activity to induce adipocyte differentiation and increase lipid synthesis in cultured animal fibroblasts or pre-adipocytes. Serotypes Ad-5, Ad-37, and particularly Ad-36 infections led to the increase of adipogenic activity in animal models of chickens (Ad-36 and -37), mice (Ad-5 and -36), rats and marmosets (Ad-36)5-8. Cytomegalovirus (CMV) as a common herpesvirus that infects over half of adults by age 40, was associated with metabolic syndrome (diagnosed as systemic inflammation, insulin resistance and gut barrier disruption etc.) particularly in females9. Recent additions to this list also include Dengue fever virus (DEGV) and treated-HIV infections (which belong to Flavivirade and retroviridae, respectively) in the patients post the viral infections entering into a latent phase, such as after antiretroviral therapy (ART) in HIV cases10-13. Notably, HIV infection itself may not cause lipohypertrophy; however, metabolic syndrome and visceral adiposity have been observed in treated patients probably due to the viral persistence and chronic inflammation resulted from antiretroviral therapy12,13. Owe to recent advancement in viral metagenomics studies (virome), it is expected that more previously unknown viruses may be identified to be “pawnobes”, which defines a responsible taxon of microbiota that subjects to adipogenic regulation. The increasing pawnobes may partly result from the selection pressure of current obesity-prone life styles in people and contemporary fattening management in livestock5,14-17. It is remarkable that most obesity-associated viruses have a livestock reservoir or share similar pathogenic persistence between livestock and people5,15-17.
Major immunological links underlying the viral infectobesity
Figure 1 illustrates most immunological links underlying obesogenesis but not limited to viral infectobesity. A majority of obesity cases are developed owing to gradual cofactorial induction that builds up immunometabolic disorders involving multiple organs in digestive (including gut microbiota), adipose, endocrine and nervous systems18-21. In the situation of viral infectobesity, excess intake of energy-dense food (such as high-fat diet, HFD) still forms a substantial physical factor for adipogenesis. Critically, active molecules, such as short- or long-chain fatty acid (LFA) that derive from diet-microbiota interaction in gut, dramatically alter immune and metabolic homeostasis locally and systemically22-24. This further leads to obesity and relevant comorbidities in involved organs (Figure 1)21,24. Major immunological links underlying obesity include: (1) local and systemic inflammation across intestine, adipose tissues, liver and even hypothalamus in the brain, (2) altered cytokine (such as TNF-α and IFNs emphasized here) and hormonal regulation, and (3) improperly activated immune cells (such as adipose macrophages and intrahepatic T cells)21-25. From an immunological view, some infections, particularly persistent viral infections as focused here, are associated and even form a reciprocal causality with obesity through their pathogenic intervention with host immune and metabolic systems at various stages of obesity development19-25. In this context, we stress the regulatory role of interferon (IFN) system in this virus-involved adipogenic process because recent studies have highlighted it as an immunological axis in the obesogenesis of viral infectobesity25-30. First, an intrinsic IFN-β signaling response to gut microbiota tone was demonstrated to be critical to maintain gut homeostasis31. A microbiota dysbiosis caused by HFD and persistent viral infections may subvert this intrinsic IFN signaling and progressively trigger low-grade gut inflammation, gut leaking and increase caloric intake and storage in both intestinal and adipose depots24,25. Second, persistent viral infections and LFA produced during adipogenesis irritate a prolonged IFN-α signaling, which has been detected in both adipose depots and fat liver during obesity development to be a biomarker for persistent viral infections and some autoimmune diseases24-30. Third, the prolonged IFN signaling contributes to the immune activation of pro-inflammatory macrophages and cytotoxic CD8+ T cells in adipose tissues and fat liver, which represent major cell markers for obesity and metabolic syndrome25. In contrast to induce effective antiviral immunity, the prolonged IFN signaling actually causes an immune dysfunction against further viral infections and aggregates to a relevant immune suppression induced by adipose cytokines (such as leptin through SOCS3). This immune deviation may form a reciprocal causality between virus susceptibility and obesity resulting in the complication of viral infectobesity7,27,30-35. In addition, chronic inflammation and immune senescence accompanying aging process generally coincide with a high incidence of viral infections in senior people36,37. It has been observed that a 5-8% higher rate of obesity in peoples of the age groups over 40 and 60 years old1,2. The potential correlation of the higher occurrence of viral infections and obesity rates in the aged groups ascribes another big topic in viral infectobesity and warrants further investigations1-3,36,37.
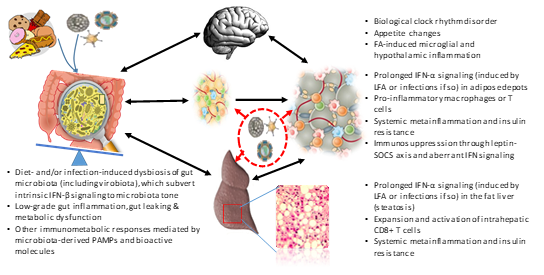
Figure 1: Schematic representation of major immunological links of cofactorial infectobesity at an inter-systemic level. While excess intake of energy-dense food (such as high-fat diet, HFD) forms a substantial physical factor for adipogenesis, active molecules derived from diet-microbiota interaction in gut, such as short- or long-chain fatty acid (LFA) in HFD, dramatically alter immune and metabolic homeostasis locally and systemically that entails obesity — a globally prevalent disease at the interface of immunity and metabolism involving multiple organs in digestive, endocrine and nervous systems. Major immunological links underlying obesity including local and systemic inflammation, altered cytokine and hormonal regulation, activated immune cells (macrophages, T cells etc.) as briefly listed by each major organ in obesogenesis. From an immunological view, some infections, particularly chronic viral infections as focused here, are associated and even form a reciprocal causality with obesity through their pathogenic intervention with host immune and metabolic systems at various stages of obesity development. Abbreviations: FA, fatty acid; IFN, interferon; PAMPs, pathogen or microbe associated molecular patterns; and SOCS3, Suppressor of cytokine signaling 3.
The metabolic basis upholding viral infectobesity
It has a chicken-or-egg disputation about infectobesity. Among a dozen of aforementioned viruses, most of them are associated with obesity and relevant morbidities through epidemiological statistics and viral coexistence (or seropositive to viral antigens) during the obesogenesis in humans and animals5-13. However, few of them have been applied to the complete tests per Koch’s postulates to establish as obesogenic pathogens38. In this context, human adenoviruses (Ad), particularly serotype Ad-36 have been well characterized in their obesogenic effect in both cell and animal models including chickens and rats5,39. The adenoviral E4orf1 protein (early region 4 open reading frame 1) was identified to be a viral mechanism mediating acute adipogenesis in both cells and animals40,41.
Indeed, transfection of E4orf1 gene alone mimicked Ad-36’s effect on the increase of glucose uptake in preadipocytes, adipocytes, or myoblasts, but reduced glucose output by hepatocytes40. Functionally acting through the Ras/PI3K signaling pathway, E4orf1 protein actually improved hyperglycemia and insulin disposal in vitro, implying a potential for anti-diabetic therapy development40,41. Peroxisome proliferator activated receptor-γ (PPARγ) is so-called a master-switch of adipocyte development. Ad-36 was capable of inducing the expression of cellular glucose transporters to facilitate energy supply and the activation of PPARγ to signal adult stem cells differentiating into adipocytes42-44. Despite the positive correlation described above, the obesogenic effect of Ad-36 was not well reproduced in mouse and monkey models5,39. Similarly, the association of Ad-36 infection with human obesity was not always significant when tested across different subject cohorts5,39. In our recent study using a rat model45, we showed that not Ad-36 alone but co-administration of Ad-36 and high-fat diet (HFD + Ad-36) induced significant obesity in rats at 8 weeks after induction. To determine what caused the difference between a previous report and ours in the obesogenic effect of Ad-36 in rat models45,46, we compared the caloric information and energy density of two diets, which were used in a previous report and our study to show difference in synergistic effect with Ad-36 in obesity induction, respectively45,46. As both diets have similar amounts of fat at ~10% in kcal, the Research Diet D12450B (Research Diets, New Brunswick, NJ) used in previous study46 that facilitated Ad-36 for obesity induction contains about 12% higher carbohydrate in kcal but 8.5% lower protein in kcal than the LabDiet5001 used in our study45. Overall, the energy density of the D12450B diet is about 0.5 kcal/g higher (3.82 vs. 3.36) than the LabDiet 5001 (Figure 2)45,46. In addition, the other components including minerals, vitamins, and fiber are similar between the two diets. Thus, this comparison implies that both the fat content and the energy density (contributed also by the other diet components, especially carbohydrate) of a diet, should be considered to uphold Ad-36 effect in obesogenic induction. This is important in terms of determining co-factorial interaction in obesity induction, indicating a certain level of energy density in a diet stands as a basis for cofactorial infectobesity at least for Ad-36 (Figure 2)45-47. Collectively, adiposity induced by energy-dense diets may set up a metabolic aberration that facilitates the virus obesogenic effect; or vice versa viruses may snatch the cellular metabolic status to benefit viral replication and exacerbates adipogenesis at the early stage of the viral infectobesity5-8,45-47. However, at the mid and late stages of obesity development, especially after the manifestation of metabolic syndrome or organ pathologies, the immunological aberration arising from adiposity and particularly persistent viral infections will play more and more role in aggravating obesity and related comorbidities as described in previous Section4,46,47.
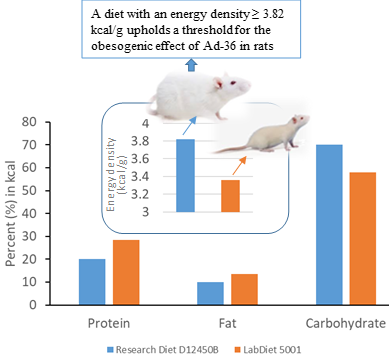
Figure 2: The high energy density of a diet upholds the viral effect in induction of viral infectobesity. Comparison of caloric information and energy density (inlet framed) of two diets that showed different in synergic effect with Ad-36 in obesity induction as in two studies, respectively45,46. As both diets have similar amount of 10% fat in kcal, the Research Diet D12450B, which showed synergistic effect with Ad-36 for obesity induction, contains about 12% higher carbohydrate in kcal but 8.5% lower protein in kcal than the LabDiet5001 that showed no synergistic effect. Overall, the energy density of the D12450B diet is about 0.5 kcal/g higher (3.82 vs. 3.36) than the LabDiet 5001. The diet formulas and contents were directly extracted and calculated from the producers’ websites at https://researchdiets.com/formulas/d12450b and https://www.labdiet.com/Products/StandardDiets/index.html.
Concluding remarks
Due to co-factorial integration in viral infections and obesity, it may be impractical to determine which factor comes first in triggering and manifesting the infectobesity5-8. I reinforce the immunometabolic links that underlying the reciprocal causality of viral infections and obesity in the infectobesity4,19-21. Along with other infectious agents beyond persistent viruses that associated with obesity, a general immunopathological common about pathogenic persistence and relevant immune deviation is a theme underlying the infectobesity4,19-21. In most cases, obesity and viral infections may interact dynamically as a reciprocal causality rather than a simple sequential relationship. Moreover, IFN (particular type I IFNs) responses have recently been postulated as a central immunological axis that governs adipocyte differentiation and T cell pathogenicity during obesity-associated metabolic diseases25,48-50. In summary, immunological dysfunction accompanying persistent viral infections and obesogenesis provides a key for understanding and potential means to treat viral infectobesity4,25,48-50.
Funding
This work was primarily supported by NIH P20-RR017686 (KSU), and in part by USDA NIFA 2018-67016-28313, USDA NIFA Evans-Allen-1013186 and NSF-IOS-1831988 to Y.S through a resource sharing plan.
Acknowledgments
The author thanks Dr. Frank Blecha and his laboratory at Kansas State University for collaborating in animal/material resources and idea discussion during the completion of relevant research projects that have generated results reviewed here. Mr. Jordan Jennings helped with critical proofreading during the revision.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Adult Obesity Facts. Available online: https://www.cdc.gov/obesity/data/adult.html (Accessed on 04-27-2019).
- Obesity. https://www.who.int/topics/obesity/en/ (Accessed on 04-29-2019).
- Ludwig DS, Friedman MI. Increasing adiposity: consequence or cause of overeating? JAMA. 2014; 311: 2168.
- Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017; 542: 177-185.
- Pasarica M, Dhurandhar NV. Infectobesity: obesity of infectious origin. Adv Food Nutr Res. 2007; 52: 61-102.
- Sohrab SS, Kamal MA, Atkinson RL, et al. Viral Infection and Obesity: Current Status and Future Prospective. Curr Drug Metab. 2017; 18: 798-807.
- Hainer V, Zamrazilová H, Kunešová M, et al. Obesity and infection: reciprocal causality. Physiol Res. 2015; 64: S105-119.
- Voss JD, Dhurandhar NV. Viral Infections and Obesity. Curr Obes Rep. 2017; 6: 28-37.
- Fleck-Derderian S, McClellan W, Wojcicki JM. The association between cytomegalovirus infection, obesity, and metabolic syndrome in U.S. adult females. Obesity (Silver Spring). 2017; 25: 626-633.
- Tan VPK, Ngim CF, Lee EZ, et al. The association between obesity and dengue virus (DENV) infection in hospitalised patients. PLoS One. 2018; 13: e0200698.
- Badawi A, Velummailum R, Ryoo SG, et al. Prevalence of chronic comorbidities in dengue fever and West Nile virus: A systematic review and meta-analysis. PLoS One. 2018; 13: e0200200.
- Lake JE. The Fat of the Matter: Obesity and Visceral Adiposity in Treated HIV Infection. Curr HIV/AIDS Rep. 2017; 14: 211-219.
- Lake JE, Stanley TL, Apovian CM, et al. Practical Review of Recognition and Management of Obesity and Lipohypertrophy in Human Immunodeficiency Virus Infection. Clin Infect Dis. 2017; 64: 1422-1429.
- Voss JD, Leon JC, Dhurandhar NV, et al. Pawnobiome: manipulation of the hologenome within one host generation and beyond. Front Microbiol. 2015; 6: 697.
- Cadwell K. The virome in host health and disease. Immunity. 2015; 42: 805-813.
- Ogilvie LA, Jones BV. The human gut virome: a multifaceted majority. Front Microbiol. 2015; 6: 918.
- Mukhopadhya I, Segal JP, Carding SR, et al. The gut virome: the 'missing link' between gut bacteria and host immunity? Therap Adv Gastroenterol. 2019; 12: 1756284819836620.
- Goodarzi MO. Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018; 6: 223-236.
- Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol. 2012; 13: 707-712.
- Harford KA, Reynolds CM, McGillicuddy FC, et al. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc. 2011; 70: 408-417.
- Francisco V, Pino J, Campos-Cabaleiro V, et al. Obesity, Fat Mass and Immune System: Role for Leptin. Front Physiol. 2018; 9: 640.
- Schroeder BO, Backhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016; 22: 1079–1089.
- Meijnikman AS, Gerdes VE, Nieuwdorp M, et al. Evaluating causality of gut microbiota in obesity and diabetes in humans. Endocr Rev. 2017; 39: 133–153.
- Sun L, Ma L, Ma Y, et al. Insights into the role of gut microbiota in obesity: pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell. 2018; 9: 397-403.
- Ghazarian M, Revelo XS, Nøhr MK, et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci Immunol. 2017; 2 pii: eaai7616.
- Fritsch SD, Weichhart T. Effects of Interferons and Viruses on Metabolism. Front Immunol. 2016; 7: 630.
- Raniga K, Liang C. Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses. 2018; 10 pii: E36.
- Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017; 38: 542-557.
- Dagenais-Lussier X, Loucif H, Murira A, et al. Sustained IFN-I Expression during Established Persistent Viral Infection: A "Bad Seed" for Protective Immunity. Viruses. 2017; 10 pii: E12.
- Wu D, Sanin DE, Everts B, et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity. 2016; 44: 1325-1336.
- Kawashima T, Kosaka A, Yan H, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-β. Immunity. 2013; 38: 1187-1197.
- Nice TJ, Robinson BA, Van Winkle JA. The Role of Interferon in Persistent Viral Infection: Insights from Murine Norovirus. Trends Microbiol. 2018; 26: 510-524.
- O'Shea D, Hogan AE. Dysregulation of Natural Killer Cells in Obesity. Cancers (Basel). 2019; 11: pii: E573.
- Terán-Cabanillas E, Hernández J. Role of Leptin and SOCS3 in Inhibiting the Type I Interferon Response During Obesity. Inflammation. 2017; 40: 58-67.
- Easterbrook JD, Dunfee RL, Schwartzman LM, et al. Obese mice have increased morbidity and mortality compared to non-obese mice during infection with the 2009 pandemic H1N1 influenza virus. Influenza Other Respir Viruses. 2011; 5: 418-425.
- Goldstein DR. Aging, imbalanced inflammation and viral infection. Virulence. 2010; 1: 295-298.
- Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016 Oct; 16(10): 626-38.
- Gradmann C. A spirit of scientific rigour: Koch's postulates in twentieth-century medicine. Microbes Infect. 2014; 16: 885-892.
- Voss JD, Atkinson RL, Dhurandhar NV. Role of adenoviruses in obesity. Rev Med Virol. 2015; 25: 379–387.
- Dhurandhar EJ, Dubuisson O, Mashtalir N, et al. E4orf1: a novel ligand that improves glucose disposal in cell culture. PLoS One. 2011; 6: e23394.
- Dhurandhar NV. Insulin sparing action of adenovirus 36 and its E4orf1 protein. J Diabetes Complications. 2013; 27: 191–199.
- Dubuisson O, Dhurandhar EJ, Krishnapuram R, et al. PPARgamma-independent increase in glucose uptake and adiponectin abundance in fat cells. Endocrinology. 2011; 152: 3648–3660.
- Floyd ZE, Stephens JM. Controlling a master switch of adipocyte development and insulin sensitivity: covalent modifications of PPARγ. Biochim Biophys Acta. 2012; 1822: 1090–1095.
- Weber KJ, Sauer M, He L, et al. PPARγ Deficiency Suppresses the Release of IL-1β and IL-1α in Macrophages via a Type 1 IFN-Dependent Mechanism. J Immunol. 2018; 201: 2054-2069.
- Sang Y, Shields LE, Sang ER, et al. Ileal transcriptome analysis in obese rats induced by high-fat diets and an adenoviral infection. Int J Obes (Lond). 2019 Jan 22. doi: 10.1038/s41366-019-0323-2. [Epub ahead of print]
- Pasarica M, Shin AC, Yu M, et al. Human adenovirus 36 induces adiposity, increases insulin sensitivity, and alters hypothalamic monoamines in rats. Obesity (Silver Spring). 2006; 14: 1905–1913.
- Hall KD. A review of the carbohydrate-insulin model of obesity. Eur J Clin Nutr. 2017; 71: 323-326.
- McGillicuddy FC, Chiquoine EH, Hinkle CC, et al. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J Biol Chem. 2009; 284: 31936-31944.
- Wieser V, Adolph TE, Grander C, et al. Adipose type I interferon signalling protects against metabolic dysfunction. Gut. 2018; 67: 157-165.
- Ying W, Kanameni S, Chang CA, et al. Interferon tau alleviates obesity-induced adipose tissue inflammation and insulin resistance by regulating macrophage polarization. PLoS One. 2014; 9: e98835.