
Implications of the HDAC6-ERK1 feed forward loop in immunotherapy
Jheng-Yu Wu1, 2, Niko Moses1,2, Wenlong Bai3, Xiaohong Mary Zhang1,2*
1Department of Oncology, Molecular Therapeutics Program, Karmanos Cancer Institute, Detroit, Michigan 48201
2Wayne State University School of Medicine, Detroit, Michigan 48201
3Department of Pathology and Cell Biology, Morsani College of Medicine, University of South Florida, Tampa, FL 33612
Abstract
The oncogene HDAC6 controls numerous cell processes that are related to tumorigenesis and metastasis, and has recently arisen as a target to treat malignancies. The ERK cascade is a classic pathway driving oncogenesis, and the components of this pathway are either highly mutated in cancers or are vital in cancer’s pathological activity. The interactions between these important components of tumor proliferation have been examined, and our research has demonstrated that they regulate each other as evidenced by different posttranslational modifications. Preclinical evidence also supports clinical trials cotargeting these two pathways, which may provide better efficacy than single treatment. Furthermore, HDAC6 and ERK both participate in the regulation of T cell maturation and may have implications on the functions of immune cells. This leads to the possibility of connecting HDAC6 and ERK to immunotherapy. In this review, we summarize the published studies about the interaction of HDAC6 and ERK cascade and their relationship to cancers. We also include the association of HDAC6 and ERK to immune system and discuss the plausibility of linking these to immunotherapy.
Introduction
Histone deacetylase 6 (HDAC6), a class IIb HDAC, plays significant roles in numerous biological and pathological processes, including cell mobility, DNA damage response, chemoresistance, transformation, and cell proliferation. Notably, the oncogenic role HDAC6 plays in human tumorigenesis makes it an emerging molecular target for cancer treatment1. The mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway is a vital signal cascade governing cell proliferation, differentiation, growth, and division2. The components of this pathway have long been targeted to treat cancer. Our previous study has shown that the ERK signaling cascade regulates the enzymatic activities of HDAC63, and several lines of evidence from our lab and other researchers’ indicate that the elements of the ERK cascade, EGFR, Ras, and ERK1/2, can be manipulated by HDAC6 via deacetylation4-6. Herein, we have reviewed the literature on HDAC6 and the ERK cascade and highlighted the main interactions and regulations between them with an emphasis on the emerging role of HDAC6 in immune checkpoint blockade.
HDAC6, ERK cascade, and cancer
Harboring two consecutive catalytic domains and two nuclear export signal, HDAC6 shows distinct properties among eleven Zn2+-dependent HDACs. Although a few reports exist showing HDAC6 in the nucleus, HDAC6 exerts the majority of its activity in the cytoplasm and most of HDAC6’s substrates are cytoplasmic proteins, such as α-tubulin, cortactin and β-catenin7. HDAC6 has been linked to different types of malignancies through overexpression and the regulation of transformation and cell migration.
Higher HDAC6 mRNA expression levels have been shown in oral squamous cell carcinoma cells8. This also occurs in acute myeloid leukemia and the leukemia cell lines HL60, K562, and KG1a9. In addition, HDAC6 mRNA overexpression has also been detected in numerous primary tumors such as pancreatic, ovarian, breast, brain, and colon cancers10. For example, HDAC6 mRNA overexpression is detected in clinical samples of ovarian carcinoma as well as ES-2, SKOV-3, and TOV-21G ovarian cancer cells.
HDAC6 exhibits the ability to promote cell migration via deacetylation of its cytoskeletal substrates α-tubulin and cortactin11,12. Studies showed that the levels of HDAC6 expression and cytoskeletal substrate’s acetylation are related to metastasis1. By examining SKOV3, MCF7, and SKBR3 cell lines, HDAC6 was discovered to play an essential role in carcinogenesis13. Moreover, HDAC6 interacts with Ku70 and Tat, suggesting a significant role in regulating antigens in tumor cells7.
Many lines of evidence have supported the notion that the ERK pathway is essential for tumor growth, and the mutational activation of different ERK elements has been revealed in numerous cancers including lung cancer, melanoma, and colon cancer14. Multiple mutations in EGFR have been reported in lung tumors, including somatic and secondary resistance mutations15. Somatic mutations such as G719S, L858R, and L861Q in exons 19 and 21, and deletions in exon 19 have been reported in patient samples and cancer-derived cell lines. In patients harboring resistance to tyrosine kinase inhibitors (TKIs) the T790M mutation has been detected, which enhances EGFR’s binding affinity for ATP and therefore decreases TKI’s efficacy. In addition to these major mutations, many other mutations throughout exons 18-21 of the EGFR tyrosine kinase domain have been discovered, correlating to TKI resistance15.
In comparison to EGFR, Ras mutations are even more frequently detected in tumors. Overall, more than 30% of cancers harbor Ras mutations, such as 61.2 (Glutamine to Arginine) and 12.2 (Glycine to Valine), which lead to persistent binding to GTP or resisting hydrolysis of GTP. These mutants impart constitutive activation upon Ras, allowing it to signal to its downstream targets inexhaustibly14,16. Drugs targeting Ras such as farnesyltransferase inhibitors were developed, but they failed clinically due to high toxicity and low specificity16.
Downstream of Ras, BRAF has been shown to prompt tumorigenesis and angiogenesis, and its active form is capable of inducing transformation in melanoma17,18, promoting invasion19. Consistently, BRAF mutations have been reported in cancers with papillary thyroid and colon cancers, harboring 50% and 10% mutation cases, respectively20,21. Other cancers, such as lung, ovarian, and breast cancers, have also been reported to contain BRAF mutations21.
Further downstream in the ERK pathway is MEK, which is also potentially associated with cancer development22. Overexpressed MEK induces NIH3T3 cell transformation23. Moreover, growth factor-stimulated differentiation and proliferation can be blocked by expression of kinase-dead mutant MEK1 in PC12 cells and in cells transformed by v-src and Ras23. So far, no evidence has indicated that MEK mutations are directly related to malignancy occurrence, but some studies have revealed that MEK mutations may confer resistance to MEK inhibitors24. This phenomenon was also reported by Emery et al. in melanoma patients that relapsed after treatment with AZD6244, a highly selective, non-ATP-competitive MEK inhibitor25.
As the critical component of the ERK pathway, ERK1/2 have been in the spotlight for years in tumor growth studies. Although ERK1/2 are not highly mutated in cancers, for tumors resistant to RAFi and MEKi, ERK1/2 could serve as an alternative target for treating recurrent disease. Evidence has shown that activation of ERK1/2 stimulated by growth factor proepithelin promotes migration and invasion of bladder cancer cells26. A separate study demonstrated that higher ERK1/2 phosphorylation is linked to higher tumor stage and lymph node metastasis in NSCLC patients27. To date, more than 160 ERK substrates have been identified including many oncogenes, such as c-Myc, ELK1, and c-Fos28. Taken together, these studies indicate that inhibition of ERK1/2 should be pursued as a target in strategies for treating malignancy.
Interactions between HDAC6 and the ERK pathway
In general, the signal transduction of the canonical ERK pathway is dominated by phosphorylation. However, other posttranslational modifications have been reported recently in manipulating the activity of ERK pathway components. Among these discoveries, HDAC6 has been revealed to associate with EGFR, Ras and ERK (Table 1). The linkage between HDAC6 and EGFR has been documented in two reports. Deribe et al., described HDAC6 as an operator of EGFR’s trafficking via modulation of microtubule acetylation status29. Reducing HDAC6 protein levels enhances EGFR intracellular trafficking and degradation. The same study has also shown that EGFR phosphorylates HDAC6 on tyrosine 570, which is within its DAC2 domain. The phosphorylation attenuates the enzymatic activity of HDAC6 that results in increasing α-tubulin acetylation and changing EGFR trafficking. Hence, these two proteins form a feedback mechanism29. A second study has further proved that HDAC6 inhibits EGFR endocytic trafficking and degradation via reduction of microtubule acetylation30. Since HDAC6 has been characterized in regulating growth factor-induced endocytosis31, like EGFR HDAC6 has also been reported to manipulate membrane protein Notch3's trafficking and lysosomal degradation via control of α-tubulin deacetylation32.
Table 1: Interaction between HDAC6 and ERK pathway components
| ERK pathway Components | Interaction | Function | References |
| EGFR | HDAC6 attenuates EGFR endocytic trafficking and degradation via deacetylating microtubules | Inhibition HDAC6 enhances EGFR degradation | Deribe et al., 2009; Gao et al., 2010 |
| EGFR | EGFR phosphorylates HDAC6 Tyr570 | EGFR phosphorylates HDAC6 and reduces HDAC6 deacetylase activity | Deribe et al., 2009 |
| KRAS | HDAC6 deacetylates KRAS | Inhibition of HDAC6 weakens cell transformation by mutational KRAS | Yang et al., 2013 |
| ERK1 | ERK1 phosphorylates HDAC6 Thr1031 and Ser1035 | Phosphorylation of HDAC6 Ser1035 is critical for its deacetylase activity | Williams et al., 2013 |
| ERK1 | HDAC6 deacetylates ERK1 Lys72 | Acetylation of ERK1 Lys72 impairs its Kinase activity | Wu et al.,2018 |
In 2012, Yang et al., reported that Ras is an acetylated protein, and Lysine 104 of K-Ras was established as an acetylation site via mass-spectrometry and mutagenesis studies. The acetylation status of this Lysine controlled K-Ras’ oncogenic activity. According to the point mutation study, the acetylation-mimetic K104Q attenuated K-Ras4B’s activity on proliferation and clonogenic survival of NIH3T3 cells33. In the following year, the same group further identified that both HDAC6 and SIRT2 as the deacetylases regulating K-Ras acetylation and oncogenic activity34.
Our studies published in 2013 have shown that ERK1 phosphorylates HDAC6 and influences HDAC6’s activity in cell migration (Fig.1)35. In the studies, we identified three possible ERK1/2 phosphorylation sites. Two of them, Threonine 1031 and Serine 1035, were identified by mass spectrometry, and Serine 1045 phosphorylation site was putatively foreseen according to the sequence of ERK1/2 recognition motif. Serine 1035 and Threonine 1031 were further confirmed as the major and minor phosphorylation site, respectively. The results also demonstrated that the non-phosphorylation mimetic mutant of HDAC6 —Alanine substitution of Serine 1035— exhibited lower tubulin deacetylase (TDAC) activity in vivo and a reduced ability to promote cell migration compared with the wild-type. A later study from Lim et al. had a conflicting conclusion, showing that an epinephrine analog, isoproterenol, increases HDAC6 mRNA and protein level via dysregulation of ERK signaling36. However, direct evidence to prove how isoproterenol manipulate ERK and HDAC6 interplay as well as results from different cell lines is warranted.
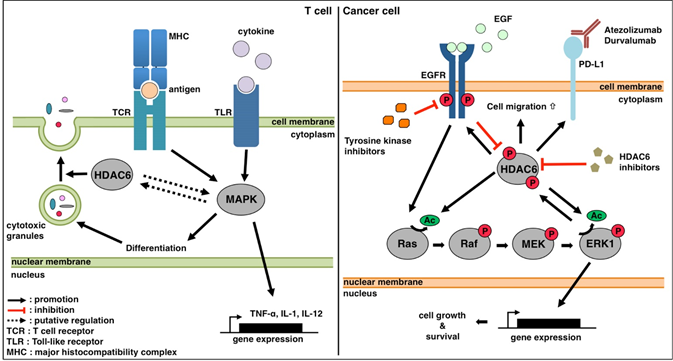
Figure 1: Working models of the interaction between HDAC6 and MAPKs in the T cell and cancer cell. Please see the text for details.
A recently released report from our lab has demonstrated that ERK1/2 are acetylated proteins, and HDAC6 and CBP/p300 are the modulators for ERK1/2 acetylation37. In this publication, we showed that ERK1/2 could be acetylated by CBP and p300 in vitro and in vivo, and both pharmacological inhibition and genetic knockdown/knockout results indicated that HDAC6 could serve as a deacetylase for ERK1/2. Additional assays utilizing inhibition and knockdown of HDAC6 confirmed that it deacetylated ERK1 in vitro and in vivo. We also examined the effect of acetylation on ERK1’s enzymatic activity. Recombinant ERK1, purified from HDAC6 knockout cells or acetylated in vitro by CBP, exhibited lower kinase activity compared with the recombinant ERK1 protein purified from control cells, revealing that acetylated ERK1 has decreased enzymatic activity (Fig.1). We later identified a novel acetylation site Lysine 72 in ERK1. Further investigations showed that the acetylation mimetic mutant K72Q displayed a reduced enzymatic activity via in vitro kinase and ELK1 reporter luciferase assays, suggesting that this mutant harbors attenuated enzymatic activity in regulating downstream transcriptional regulator ELK1. Structural analyses demonstrated that the acetylation mimetic mutant K72Q, but not the deacetylation mimetic mutant K72R, prevents the formation of a salt bridge with Aspartic Acid 117 and a linkage to Tyrosine 119 with a hydrogen bond. This obstruction causes the lower stability of the β3-strand, reduced ATP binding, and further lowered ERK1’s enzymatic activity.
The intimate interaction between HDAC6 and ERK pathway (Table 1) makes therapeutic targeting HDAC6 a new option to treat cancers hosting destructive ERK pathway components. According to the above two studies from our group, HDAC6 and ERK1 regulate each other in a positive manner; ERK1 promotes HDAC6’s tubulin deacetylase activity, and HDAC6 increases ERK1’s kinase activity. Based upon these observations, we hypothesized that these two proteins form a positive feed-forward loop to enhance oncogenic activity in cancers, making these two proteins suitable for simultaneous targeting to interrupt tumor growth.
Implications of co-targeting HDAC6 and the MAPK pathway in immune checkpoint therapy
Roles of the ERK pathway and HDAC6 in the Immune Response
When implementing combination therapies in the clinic, a factor that must be taken into account in the modern era of oncology is the impact of these therapeutics on the immune response. As our understanding of the immune system evolves, oncologists are beginning to realize that the long-term antitumor responses patients have achieved after chemotherapeutic regimens may be due to the presentation of tumor-specific antigens to the immune system after chemotherapy-induced tumor cell lysis38. To induce long-term responses in a larger proportion of patients, timing of therapeutics and which factors to target must be optimized to minimize the immunosuppressive features of chemotherapy while maximizing the tumor cell killing potential of the combination to allow the immune system access to the repertoire of tumor antigens and the ability to amount an immune response against the tumor. To accomplish this aim, the role of the ERK pathway and HDAC6 in the immune response must be considered.
The MAPK pathway is an important facet of both innate and adaptive immunity. In the innate arm of the immune system, MAPK participates in signaling downstream of receptors in innate immune cells. Toll-like receptors (TLRs) are the most prominent family that signal via the MAPK pathway. There are 11 mammalian TLRs, each of which recognizes specific microorganism molecular patterns foreign to the host. Upon activation of a TLR, adaptors are recruited which further activate MAP kinases p38, JNK, and ERK1/2 (Fig.1). Recruitment of any of the three MAP kinases leads to activation of transcription factor AP-1, which induces the transcription of type I interferons, inflammatory cytokines, and chemokines. Activation of AP-1 is the main role that ERK1/2 plays in the immune response, while p38 and JNK have additional roles which will be described below.
The particular adaptors that mediate recruitment and the specific cytokines and chemokines produced are cell-type specific. Macrophages express TLR2 and TLR4, which recognize peptidoglycans and LPS, respectively39,40. If either of these receptors is engaged, adaptor protein MyD88 will recruit a signalsome (IRAK, TRAF6, ESCIT) to mediate activation of the MAPK pathway41, which will induce the production of inflammatory cytokines (e.g. TNF-α, IL-1, IL-12)42. Recognition of viral dsRNA by NK cells occurs through TLR3, which induces the recruitment of the TRIF/TICAM-1 adaptor complex43. The subsequent signaling cascade activates p38 and leads to transcription of IFN-γ and chemokines CXCL-10 and -843. In the adaptive arm of the immune system, MAPK plays a critical role in T cell differentiation by participating in the ITAM signaling pathway. When a naïve T cell engages its TCR with antigen-loaded MHC and its CD28 is bound by B7, cytosolic adaptors SLP76 and Grab2 will be recruited to the complex of Syk kinases, and LAT adaptors present at the intracellular portion of the TCR. This signaling complex activates sos-ras-MEK-ERK44. This cascade results in the recruitment of transcription factor AP-1, which activates a mature T cell transcriptional profile consisting of type I interferons, chemokines, and inflammatory cytokines45. In activated CD4+ and CD8+ T cells, p38 has been shown to regulate the production of antitumor cytokine IFN-γ, although this pathway has been better characterized in the CD4+ helper T cell population46. The MAPK pathway also participates in the termination of adaptive immune response; JNK has been reported to regulate apoptosis in T cells47, and activation of p38 can induce apoptosis in CD8+ T cells and serve as a negative feedback loop to the aforementioned role it plays in IFN-γ production in T cells48.
Like the MAPK signaling pathway, HDAC6 also functions in both the innate and adaptive arms of the immune system. However, while MAPK mainly serves to transduce signals from engaged immune receptors to activate particular transcriptional profiles, HDAC6 mainly acts at the posttranslational level via its canonical targets (i.e. Hsp90 and α-tubulin). HDAC6 is an appealing target for antitumor therapy because it is upregulated in a variety of tumor types, including breast and ovarian cancer, glioblastoma, and AML1. Despite the current utilization of HDAC6-specific inhibition in the clinic for cancer treatment, mechanistic studies of HDAC6’s immune activity propose using HDAC6 inhibition in autoimmune disease, not cancer49. For instance, HDAC6 can regulate the acetylation of critical regulatory T cell (Treg) transcription factor FoxP3, and deacetylated FoxP3 is more likely to be ubiquitinated and degraded via the proteasome pathway. Consistent with this role, both genetic ablation and pharmacological inhibition of HDAC6 can increase the suppressive function of Tregs in vivo50,51. Inhibition of HDAC6 with Tubastatin A can also drastically reduce production of TNF and IL-6 in the THP1 macrophage cell line52. In murine models, HDAC6 expression is enhanced specifically in the CD8+ subset of T cells upon activation with anti-CD3/CD28 antibody49. HDAC6 also controls the formation of an immune synapse between antigen presenting cells (APCs) and effector T cells via its deacetylase activity on the α-tubulin cytoskeleton; HDAC6 was found to concentrate at the contact point between these two cell types53. However, this study by Sanchez-Madrid and colleagues also proposed a role for HDAC6 in termination of the immune synapse, as overexpressed HDAC6 may promote synapse termination by internalizing the T cell receptor (TcR). In CD8+ cytotoxic T cells, HDAC6 also mediates the response to TcR engagement by APCs; when a T cell recognizes an antigen on a target cell, HDAC6 mediates the transport of toxic granules along the cytoskeleton to the plasma membrane to kill the target (Fig.1)54.
In the innate arm of the immune system, HDAC6 has been found to control LPS-induced activation of macrophage and their subsequent production of pro-inflammatory cytokines55. In response to bacterial infection, HDAC6 facilitates bacterial autophagy by dendritic cells and maintains proper activation of TLR signaling in these cells56. Intriguingly, these researchers also found that the bone marrow derived dendritic cells (BMDCs) obtained from HDAC6 KO mice had reduced phosphorylation of MAPK signaling components in response to bacterial infection56. While these observations presented a new role for HDAC6 in the clearance of bacterial, HDAC6 has long been established as serving an antiviral role in host cells; HDAC6 upregulates IFN-β production in the host in response to viral infection57, promotes activity of RNA-virus sensor RIG-I58, and HDAC6 knockout has increased murine susceptibility to viral infection in multiple studies59,60.
Both ERK1 and HDAC6 appear to play pro-immunogenic roles, as evidence by the breadth of literature discussed above, which should give oncologists pause when considering whether inhibition of either of these enzymes could pair with immune checkpoint blockade. In order to successfully combine ERK1 inhibition, HDAC6 inhibition, and immunotherapy, a great deal of thought in terms of method of administration, dose, and dose timing will be critical to achieve efficacy.
Relevance of HDAC6 Activity in Immune Checkpoint Therapy
The recent advances made in the clinic in terms of checkpoint blockade inhibitor combination therapy are what renowned immunologist Dr. Padmanee Sharma has referred to as “hypothesis generating”; synergy observed in these clinical combinations must eventually be brought back to the bench to determine the mechanisms of synergy, which can offer information about alternative approaches for future clinical trials. The aforementioned combinations of epigenetic modifiers and immune checkpoint blockade inhibitors are generating hypotheses, but the mechanisms behind the efficacy of these treatments remain to be elucidated. We have an understanding of the roles that the ERK1 pathway and HDAC6 play in the immune response, but the facets that are involved in an improved patient response to immune checkpoint blockade are as of yet undetermined.
It bears mentioning that the rationale behind pairing pan-HDAC inhibitors with immune checkpoint blockade differs greatly from the targeting of HDAC6 alone. Pan-HDAC inhibitors and HDAC class I specific inhibitors have defined pro-immunogenic functions; increased MHC I and TAP expression on the surface of tumors61, increased production of tumor-associated antigens62, and increased chemokine expression in T cells63 to name a few. However, pan- and class I-HDAC inhibition involves more severe side effects than HDAC6-specific inhibition, including gastrointestinal dysfunction, lowered blood platelet levels, risk of hemorrhage, fatigue, and potential for significant cardiac toxicity64. If therapeutic efficacy is possible with HDAC6 inhibition alone, isoform-specific inhibition in lieu of pan-HDAC inhibition is expected to provide better safety profiles and therapeutic indexes than the pan inhibitors that have been approved by the FDA. For instance, Acetylon Pharmaceuticals has developed HDAC6-specific inhibitor ricolinostat (ACY-1215), which is currently in phase II clinical trials for multiple myeloma and lymphoma. Ricolinostat is dosed more frequently and better tolerated than panobinostat or vorinostat and has the potential to be combined with immune checkpoint blockade65,66.
Despite the majority of mechanistic studies indicating that HDAC6 plays a critical role in the proper function of the immune system, this has not stopped the utilization of HDAC6 inhibitors in combination with immunotherapy both preclinically and clinically. HDAC6 inhibitors are currently tested in the clinic paired with nivolumab in phase I trials against NSCLC and multiple myeloma. HDAC6 inhibitor ACY-241 has recently been combined with α-PD-L1 in a preclinical multiple myeloma model, where enhanced antitumor cytotoxicity was observed67. In melanoma cell lines, specific targeting of HDAC6 induced G1 arrest and upregulated tumor-associated antigens (TAAs) and MHC class I, reducing tumor growth and increasing immunogenicity in this model68. This group also tested primary melanoma samples and found that inhibition of HDAC6 downregulated PD-L1 on the surface of these cells and impaired xenograft growth69. In a preclinical model of NSCLC, HDAC6 inhibition with ricolinostat enhanced activation of T cells via improved function of antigen presenting cells. Combining ricolinostat with bromodomain inhibitor JQ1 further improved the immunogenicity of the tumor microenvironment by abrogating FoxP3+ regulatory T cell suppressive activity, and all of these effects in combination have led to immune-mediated growth arrest of tumors and extended the survival of mice70.
The stark contrast between the success of clinical and preclinical combination therapy studies of HDAC6-specific inhibition with immunotherapy and our current understanding of HDAC6’s role in mediating the therapeutic efficacy underlies a need for more research into HDAC6’s role in the adaptive arm of the immune system. It is still too soon to reject the combination of HDAC6 inhibition with immunotherapy; one needs only to look at the history of PD-1 to understand why initial studies are not always predictive of future clinical relevance. PD-1 was first discovered in 1992, but anti-PD-1 therapeutics did not receive FDA approval until 2014 (pembroluzimab). In the interim, there were numerous studies suggesting that PD-1 targeting would not be useful in the clinic. The first transgenic PD-1-/- mice had an increased incidence of spontaneous lupus-like autoimmune disorders71,72, indicating that PD-1 targeting would harm patients. When examining PD-1 ligand PD-L1 in vitro, researchers found that both human and mouse cultured tumor lines had almost no expression of PD-L1 on their surface73. However, clinical relevance came to light when freshly isolated patient tumors were found to express high levels of PD-L1, which was due to IFN-γ-induced upregulation of the ligand74. While the current mechanistic studies have revealed why targeting HDAC6 may not benefit cancer patients, the successful clinical trials combining HDAC6 and immune checkpoint blockade indicate that there is a pro-immunogenic role for HDAC6 inhibition that remains to be realized.
Concurrently targeting HDAC6 and ERK pathway
The rationale behind combination therapy is to overcome drug resistance and avoid malignancy metastasis, and this dual target strategy can exploit inhibition or modulation of traditional cancer markes such as HDACs, kinases, or apoptotic proteins75. The close connection between HDAC6 and the components of the ERK pathway leads to the strategy of concurrent targeting of HDAC6 and the ERK pathway in cancer. Simultaneous inhibition of HDACs and ERK pathway components has been researched in several cell line studies and showed synergistic effect7. Other reports have shown that inhibiting HDACs might benefit the TKI’s effect76,77.
To aim not just a single target but more than two objectives at the same time for better consequence, the approach applying the dual-target/multi-target agent to treat malignancies has arisen78,79. Few years ago, a multitarget inhibitor CUDC-101 was developed to target class I and class II HDACs, EGFR, and HER2 simultaneously. This multifunctional compound, compared to HDACi and TKI combination treatment, displayed enhanced efficacy via inhibition of cell growth across a board range of cell lines, including NSCLC, sarcoma, and cancers of breast, prostate, colon, liver, etc80. It also exhibited effective growth inhibition against various xenograft tumors such as liver cancer, breast cancer, NSCLC, head and neck squamous cell carcinoma (HNSCC), and pancreatic cancer81. Same group further showed CUDC-101’s capability of inhibiting migration and invasion in multiple cell lines including erlotinib-resistant HCC82782. These studies support co- targeting HDACs and the MAPK pathway components.
The third generation TKI osimertinib (AZD9291) was recently developed for overcoming resistance to first and second generation TKIs in malignancy, especially NSCLCs with the EGFR T790M mutant83. In a clinical trial, osimertinib showed better efficacy in untreated advanced NSCLC patients with mutant EGFR than the standard EGFR-TKI and had a similar safety profile as early generation TKIs84. In TKI-pretreated NSCLC patients harboring the T790M mutant, osimertinib also caused higher objective responsive rate, durable response, and encouraging progression-free survival85. Combination of TKI and immune checkpoint blockade has entered clinical trials. These trials, including different combination such as nivolumab/erlotinib, osimertinib/durvalumab, atezolizumab/erlotinib, and gefitinib/durvalumab showed encouraging anti-cancer activity. However, these cotreatments also caused severe side effects such as interstitial lung disease86. Aside from these studies, more trials using both TKIs and immunotherapy are ongoing or will open87. Although the trials showed the toxicity, the effective clinical activity makes the combination of EGFR TKI and immunotherapy a promising strategy to manage advanced NSCLC.
Several clinical trials involving the combination of epigenetic or posttranslational modification modulators with immune checkpoint inhibitors are either recruiting or going to be initiated88. Among these clinical trials, most of them included nonselective pan-HDAC inhibitors (panobinostat, entinostat, and vorinostat) with anti-immune checkpoint drugs (pembrolizumab, ipilimumab, atezolizumab, and nivolumab) for treating different cancer types. However, four FDA-approved HDAC inhibitors, panobinostat, belinostat, romidepsin, and vorinostat, are all pan-HDAC inhibitors89. Among these non-selective inhibitors, romidepsin and vorinostat, the first two approved drugs, showed sever limits such as inefficacious and high drug-induced side effect in solid tumor90. Singly targeting at an HDAC is a feasible way for leading to fewer adverse reactions and better clinical usefulness. As the previously mentioned discussion, HDAC6 is now a novel target for treating malignancies and several HDAC6-specific inhibitors have been developed91. Applying the HDAC6-specific inhibitors in the combination therapy with anti-immune checkpoint drugs could show promising expected result. A planned trial has been designed using ACY-241 with nivolumab and ipilimumab in a group of unresectable NSCLC88. The result of this trial could validate the concept of cotargeting HDAC6 and immune checkpoint components. The other study has shown that cotreatment of pan-HDACi inhibitor ITF-2357 and DNA methyltransferase inhibitors azacitidine lowered MYC-medicated cell proliferation via immune signaling enhancement92. This study also support our hypothesis, but further study with HDAC6 specific should be warranted.
Conclusions and future challenges
As the main growth-driver in numerous cancer types, the ERK pathway is considered a key target for cancer treatment. However, acquired or developed resistance to current inhibitors is almost inevitable2. A similar situation is also applicable for HDAC inhibitors, and the mechanisms of resistance include the activation of p21, increased expression of Bcl-2, and induction of ERK pathway93. Furthermore, constitutive expression of MEK1 in lung cancer cell lines diminishes the cytotoxicity generated by pan-HDAC inhibitor romidepsin93, suggesting that combination therapy may offer a promising approach to avoid resistance to single agent treatment. Nevertheless, the detailed mechanisms of how these cotreatments function requires further definition. The combination of HDAC inhibitors or TKIs with immunotherapy has emerged recently, and these treatment regimens have produced encouraging clinical outcomes. Although these promising results make combination therapies an appealing choice for patients, further investigation is necessary to reveal the underlying mechanisms for the purpose of improving efficacy while reducing the toxicity to patients. Studies specifically designed to focus on how the HDAC6-ERK interaction impacts the immune system, and whether the feed-forward loop is pro- or anti-immunogenic, could be carried out. Because HDAC6 KO mice are readily available, we may consider implementing ERK pathway inhibitors and immune checkpoint blockade in HDAC6 KO mice to uncover the mechanism of interplay between these components. The knockout mice would also be useful for determining how critical HDAC6 is for cytotoxic T cell function; T cells derived from both HDAC6 KO and HDAC6 WT mice could be treated with CD3/CD28 antibody, and activation assessed via ELISPOT for IFN-γ production. Although the effect of HDAC6 or ERK in the immune system has been studied, the co-effect of these two proteins on immune cells is still not clear. It would be worth trying co-treat the immune cells to understand the joint effect from these two proteins.
Acknowledgements
This work was supported in part by National Institutes of Health Grant R01CA164147 and by Karmanos Cancer Institute start-up funds (to X. M. Z).
Conflicts of Interest
All authors certify that they have no conflicts of interest to disclose.
References
- Aldana?Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011; 2011: 875824. doi:10.1155/2011/875824
- Samatar AA, Poulikakos PI. Targeting RAS?ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014; 13: 928?942. doi:10.1038/nrd4281
- Williams KA, Zhang M, Xiang S, et al. Extracellular signal?regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem. 2013; 288: 33156?33170, doi:10.1074/jbc.M113.472506
- Deribe YL, Wild P, Chandrashaker A, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal.2009; 2: ra84. doi:10.1126/scisignal.2000576
- Yang MH, Laurent G, Bause AS, et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K?RAS. Molecular cancer research : MCR. 2013; 11: 1072?1077. doi:10.1158/1541?7786.mcr?13?0040?t
- Wu JY, Xiang S, Zhang M, et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal? regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J Biol Chem. 2018; 293: 1976?1993. doi:10.1074/jbc.M117.795955
- Haakenson J, Wu JY, Xiang S, et al. HDAC6?Dependent Functions in Tumor Cells: Crossroad with the MAPK Pathways. Crit Rev Oncog. 2015; 20: 65?81.
- Sakuma T, Uzawa K, Onda T, et al. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int J Oncol. 2006; 29: 117?124.
- Bradbury CA, Khanim FL, Hayden R, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005; 19: 1751?1759, doi:10.1038/sj.leu.2403910.
- de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003; 370: 737?749. doi:10.1042/BJ20021321
- Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule?associated deacetylase. Nature. 2002; 417: 455?458. doi:10.1038/417455a
- Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Molecular cell. 2007; 27: 197?213. doi:10.1016/j.molcel.2007.05.033
- Lee YS, Lim KH, Guo X, et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008; 68: 7561?7569. doi:10.1158/0008? 5472.CAN?08?0188
- Roberts PJ, Der CJ. Targeting the Raf?MEK?ERK mitogen?activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26: 3291?3310. doi:10.1038/sj.onc.1210422
- da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011; 49?69. doi:10.1146/annurev?pathol?011110? 130206
- McCormick F. c?Raf in KRas Mutant Cancers: A Moving Target. Cancer Cell. 2018; 33: 158?159. doi:10.1016/j.ccell.2018.01.017
- Hingorani SR, Jacobetz MA, Robertson GP, et al. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Research. 2003; 63: 5198?5202.
- Sharma A, Trivedi NR, Zimmerman MA, et al. Mutant B?V599E?RAF regulates growth and vascular development of malignant melanoma tumors. Cancer Research. 2005; 65: 2412?2421, doi:Doi 10.1158/0008?5472.Can?04?2423
- Sumimoto H, Hirata K, Yamagata S, et al. Effective inhibition of cell growth and invasion of melanoma by combined suppression of BRAF (V599E) and Skp2 with lentiviral RNAi. International Journal of Cancer. 2006; 118: 472?476. doi:10.1002/ijc.21286
- Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis ? RAF/RAS oncogenes and mismatch? repair status. Nature. 2002; 418: 934?934. doi:10.1038/418934a
- Roberts PJ, Der CJ. Targeting the Raf?MEK?ERK mitogen?activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26: 3291?3310. doi:10.1038/sj.onc.1210422
- Sebolt?Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999; 5: 810?816. doi:Doi 10.1038/10533
- Cowley S, Paterson H, Kemp P, et al. Activation of Map Kinase Kinase Is Necessary and Sufficient for Pc12 Differentiation and for Transformation of Nih 3t3 Cells. Cell. 1994; 77: 841?852. doi:Doi 10.1016/0092? 8674(94)90133?3
- Villanueva J, Infante JR, Krepler C, et al. Concurrent MEK2 Mutation and BRAF Amplification Confer Resistance to BRAF and MEK Inhibitors in Melanoma. Cell Reports. 2013; 4: 1090? 1099. doi:10.1016/j.celrep.2013.08.023
- Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B?RAF inhibition. P Natl Acad Sci USA. 2009; 106: 20411?20416. doi:10.1073/pnas.0905833106
- Monami G, Gonzalez EM, Hellman M, et al. Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex. Cancer Research. 2006; 66: 7103?7110. doi:10.1158/0008?5472.Can?06?0633
- Vicent S, López-Picazo JM, Toledo G, et al. ERK1/2 is activated in non?small?cell lung cancer and associated with advanced tumours. Brit J Cancer. 2004; 90: 1047?1052. doi:10.1038/sj.bjc.6601644
- Yoon S, Seger R. The extracellular signal?regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006; 24: 21?44. doi:10.1080/02699050500284218
- Deribe YL, Wild P, Chandrashaker A, et al. Regulation of Epidermal Growth Factor Receptor Trafficking by Lysine Deacetylase HDAC6. Sci Signal. 2009; 2. doi:ARTN ra8410.1126/scisignal.2000576
- Gao YS, Hubbert CC, Yao TP. The Microtubule?associated Histone Deacetylase 6 (HDAC6) Regulates Epidermal Growth Factor Receptor (EGFR) Endocytic Trafficking and Degradation. Journal of Biological Chemistry. 2010; 285: 11219?11226. doi:10.1074/jbc.M109.042754
- Gao YS, Hubbert CC, Lu J, et al. Histone deacetylase 6 regulates growth factor?induced actin remodeling and endocytosis. Mol Cell Biol. 2007; 27: 8637?8647. doi:10.1128/MCB.00393?07
- Pinazza M, Ghisi M, Minuzzo S, et al. Histone deacetylase 6 controls Notch3 trafficking and degradation in T?cell acute lymphoblastic leukemia cells. Oncogene. 2018. doi:10.1038/s41388?018?0234?z
- Yang MH, Nickerson S, Kim ET, et al. Regulation of RAS oncogenicity by acetylation. P Natl Acad Sci USA. 2012; 109: 10843?10848. doi:10.1073/pnas.1201487109
- Yang MH, Laurent G, Bause AS, et al. HDAC6 and SIRT2 Regulate the Acetylation State and Oncogenic Activity of Mutant K?RAS. Mol Cancer Res. 2013; 11: 1072?1077. doi:10.1158/1541?7786.Mcr?13?0040?T
- Williams KA, Zhang M, Xiang S, et al. Extracellular Signal?regulated Kinase (ERK) Phosphorylates Histone Deacetylase 6 (HDAC6) at Serine 1035 to Stimulate Cell Migration. Journal of Biological Chemistry. 2013; 288: 33156?33170. doi:10.1074/jbc.M113.472506
- Lim JA, Juhnn YS. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp Mol Med. 2016; 48: e204. doi:10.1038/emm.2015.98
- Wu JY, Xiang S, Zhang M, et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal? regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. Journal of Biological Chemistry. 2018; 293: 1976?1993. doi:10.1074/jbc.M117.795955
- Zitvogel L, Apetoh L, Ghiringhelli F, et al. Immunological aspects of cancer chemotherapy. Nature reviews. Immunology. 2008; 8: 59?73. doi:10.1038/nri2216
- Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll?like receptor 4 (TLR4)?deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of immunology (Baltimore, Md. : 1950). 1999; 162: 3749?3752.
- Yang RB, Mark MR, Gray A, et al. Toll?like receptor?2 mediates lipopolysaccharide?induced cellular signalling. Nature. 1998; 395: 284?288. doi:10.1038/26239
- Pan ZK. Toll?like receptors and TLR?mediated signaling: more questions than answers. American journal of physiology. Lung cellular and molecular physiology. 2004; 286: L918?920. doi:10.1152/ajplung.00381.2003
- Koul A, Herget T, Klebl B, et al. Interplay between mycobacteria and host signalling pathways. Nature reviews. Microbiology. 2004; 2: 189?202. doi:10.1038/nrmicro840
- Pisegna S, Pirozzi G, Piccoli M, et al. p38 MAPK activation controls the TLR3?mediated up? regulation of cytotoxicity and cytokine production in human NK cells. Blood. 2004; 104: 4157?4164. doi:10.1182/blood?2004?05?1860
- Dustin ML. T?cell activation through immunological synapses and kinapses. Immunological reviews. 2008; 221: 77?89. doi:10.1111/j.1600?065X.2008.00589.x
- Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. International immunology. 2009; 21: 317?337. doi:10.1093/intimm/dxp017
- Rincon M, Enslen H, Raingeaud J, et al. Interferon?gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. The EMBO journal. 1998; 17: 2817?2829. doi:10.1093/emboj/17.10.2817
- Dong C, Yang DD, Wysk M, et al. Defective T cell differentiation in the absence of Jnk1. Science (New York, N.Y.). 1998; 282: 2092?2095.
- Merritt C, Enslen H, Diehl N, et al. Activation of p38 mitogen?activated protein kinase in vivo selectively induces apoptosis of CD8(+) but not CD4(+) T cells. Molecular and cellular biology. 2000; 20: 936?946.
- Tsuji G, Okiyama N, Villarroel VA, et al. Histone deacetylase 6 inhibition impairs effector CD8 T?cell functions during skin inflammation. The Journal of allergy and clinical immunology. 2015; 135: 1228?1239. doi:10.1016/j.jaci.2014.10.002
- de Zoeten EF, Wang L, Butler K, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T?regulatory cells. Molecular and cellular biology. 2011; 31: 2066?2078. doi:10.1128/mcb.05155?11
- Beier UH, Wang L, Han R, et al. Histone deacetylases 6 and 9 and sirtuin?1 control Foxp3+ regulatory T cell function through shared and isoform?specific mechanisms. Science signaling. 2012; 5: ra45. doi:10.1126/scisignal.2002873
- Vishwakarma S, Iyer LR, Muley M, et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti?inflammatory and anti?rheumatic effects. International immunopharmacology. 2013; 16: 72?78. doi:10.1016/j.intimp.2013.03.016
- Serrador JM, Cabrero JR, Sancho D, et al. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity. 2004; 20: 417?428.
- Nunez?Andrade N, Iborra S, Trullo A, et al. HDAC6 regulates the dynamics of lytic granules in cytotoxic T lymphocytes. Journal of cell science. 2016; 129: 1305?1311. doi:10.1242/jcs.180885
- Yan B, Xie S, Liu Z, et al. HDAC6 deacetylase activity is critical for lipopolysaccharide? induced activation of macrophages. PloS one. 2014; 9: e110718. doi:10.1371/journal.pone.0110718
- Moreno?Gonzalo O, Ramírez-Huesca M, Blas-Rus N, et al. HDAC6 controls innate immune and autophagy responses to TLR?mediated signalling by the intracellular bacteria Listeria monocytogenes. PLoS pathogens. 2017; 13: e1006799. doi:10.1371/journal.ppat.1006799
- Nagarajan U. Induction and function of IFNbeta during viral and bacterial infection. Critical reviews in immunology. 2011; 31: 459?474.
- Choi SJ, Lee HC, Kim JH, et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG?I. The EMBO journal. 2016; 35: 429?442. doi:10.15252/embj.201592586
- Chattopadhyay S, Fensterl V, Zhang Y, et al. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF?3 are regulated by the acetylation and phosphorylation of its coactivators. mBio. 2013; 4. doi:10.1128/mBio.00636?12
- Wang D, Meng Q, Huo L, et al. Overexpression of Hdac6 enhances resistance to virus infection in embryonic stem cells and in mice. Protein & cell. 2015; 6: 152?156. doi:10.1007/s13238?014?0120?6
- Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer immunology, immunotherapy : CII. 2008; 57: 647?654. doi:10.1007/s00262?007?0402?4
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature reviews. Cancer. 2006; 6: 38?51. doi:10.1038/nrc1779
- Zheng H, Zhao W, Yan C, et al. HDAC Inhibitors Enhance T?Cell Chemokine Expression and Augment Response to PD?1 Immunotherapy in Lung Adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016; 22: 4119?4132. doi:10.1158/1078?0432.ccr?15?2584
- Subramanian S, Bates SE, Wright JJ, et al. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals (Basel, Switzerland). 2010; 3: 2751?2767. doi:10.3390/ph3092751
- Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nature reviews. Drug discovery. 2014; 13: 673?691. doi:10.1038/nrd4360
- Wang Y, Wallach J, Duane S, et al. Developing selective histone deacetylases (HDACs) inhibitors through ebselen and analogs. Drug design, development and therapy. 2017; 11: 1369? 1382. doi:10.2147/dddt.s124977
- Ray A, Das DS, Song Y, et al. Combination of a novel HDAC6 inhibitor ACY?241 and anti?PD?L1 antibody enhances anti?tumor immunity and cytotoxicity in multiple myeloma. Leukemia. 2018; 32: 843?846. doi:10.1038/leu.2017.322
- Woan KV, Lienlaf M, Perez-Villation. Molecular oncology. 2015; 9: 1447?1457. doi:10.1016/j.molonc.2015.04.002
- M L, P PV, T K, et al. Essential role of HDAC6 in the regulation of PD?L1 in melanoma. Molecular oncology. 2016; 10: 735?750. doi:10.1016/j.molonc.2015.12.012
- Adeegbe DO, Liu Y, Lizotte PH, et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non?Small Cell Lung Cancer. Cancer discovery. 2017; 7: 852?867. doi:10.1158/2159? 8290.cd?16?1020
- Nishimura H, Nose M, Hiai H, et al. Development of lupus?like autoimmune diseases by disruption of the PD?1 gene encoding an ITIM motif?carrying immunoreceptor. Immunity. 1999; 11: 141?151.
- Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD?1 receptor? deficient mice. Science (New York, N.Y.). 2001; 291: 319?322. doi:10.1126/science.291.5502.319
- Dong H, Strome SE, Salomao DR, et al. Tumor?associated B7?H1 promotes T?cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002; 8: 793?800. doi:10.1038/nm730
- Hirano F, Kaneko K, Tamura H, et al. Blockade of B7?H1 and PD?1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer research. 2005; 65: 1089?1096.
- Bayat Mokhtari R, Homayouni TS, Baluch N, et al. Combination therapy in combating cancer. Oncotarget. 2017; 8: 38022?38043. doi:10.18632/oncotarget.16723
- Edwards A, Li JN, Atadja P, et al. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor? dependent human lung cancer cells. Molecular Cancer Therapeutics. 2007; 6: 2515? 2524. doi:10.1158/1535?7163.Mct?06?0761
- Greve G, Schiffmann I, Pfeifer D, et al. The pan?HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR?mutated and ?wildtype non?small cell lung cancer cells. Bmc Cancer. 2015; 15. doi:ARTN 94710.1186/s12885?015?1967?5 (2015).
- Wu YW, Hsu KC, Lee HY, et al. A Novel Dual HDAC6 and Tubulin Inhibitor, MPT0B451, Displays Anti?tumor Ability in Human Cancer Cells in vitro and in vivo. Front Pharmacol. 2018; 9: 205. doi:10.3389/fphar.2018.00205
- Mondello P, Derenzini E, Asgari Z, et al. Dual inhibition of histone deacetylases and phosphoinositide 3?kinase enhances therapeutic activity against B cell lymphoma. Oncotarget. 2017; 8: 14017?14028. doi:10.18632/oncotarget.14876
- Lai CJ, Bao R, Tao X, et al. CUDC?101, a Multitargeted Inhibitor of Histone Deacetylase, Epidermal Growth Factor Receptor, and Human Epidermal Growth Factor Receptor 2, Exerts Potent Anticancer Activity. Cancer Research. 2010; 70: 3647?3656. doi:10.1158/0008?5472.Can?09?3360
- Cai X, Zhai HX, Wang J, et al. Discovery of 7?(4?(3?Ethynylphenylamino)?7?methoxyquinazolin?6?yloxy)?N?hydroxyheptanamide (CUDC?101) as a Potent Multi?Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry. 2010; 53: 2000?2009. doi:10.1021/jm901453q
- Wang J, Pursell NW, Samson ME, et al. Potential Advantages of CUDC?101, a Multitargeted HDAC, EGFR, and HER2 Inhibitor, in Treating Drug Resistance and Preventing Cancer Cell Migration and Invasion. Molecular Cancer Therapeutics. 2013; 12: 925?936. doi:10.1158/1535?7163.Mct?12?1045
- Zhou WJ, Ercan D, Chen L, et al. Novel mutant?selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009; 462: 1070?1074. doi:10.1038/nature08622
- Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in Untreated EGFR?Mutated Advanced Non?Small? Cell Lung Cancer. New Engl J Med. 2018; 378: 113?125. doi:10.1056/NEJMoa1713137
- Yang JCH, Ahn MJ, Kim DW, et al. Osimertinib in Pretreated T790M?Positive Advanced Non? Small?Cell Lung Cancer: AURA Study Phase II Extension Component. Journal of Clinical Oncology. 2017; 35: 1288?+. doi:10.1200/Jco.2016.70.3223
- Ahn MJ, Sun JM, Lee SH, et al. EGFR TKI combination with immunotherapy in non?small cell lung cancer. Expert Opin Drug Saf. 2017; 16: 465? 469. doi:10.1080/14740338.2017.1300656
- Moya?Horno I, Viteri S, Karachaliou N, et al. Combination of immunotherapy with targeted therapies in advanced non?small cell lung cancer (NSCLC). Ther Adv Med Oncol. 2018; 10. doi:10.1177/1758834017745012
- Mazzone R, Zwergel C, Mai A, et al. Epi?drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics. 2017; 9: 59. doi:10.1186/s13148?017?0358?y
- Halsall JA, Turner BM. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays. 2016; 38: 1102?1110. doi:10.1002/bies.201600070
- Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012; 4: 505?524. doi:10.4155/fmc.12.3
- Wang XX, Wan RZ, Liu ZP. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur J Med Chem. 2018; 143: 1406?1418. doi:10.1016/j.ejmech.2017.10.040
- Topper MJ, Vaz M, Chiappinelli KB, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017; 171: 1284?1300 e1221. doi:10.1016/j.cell.2017.10.022
- Robey RW, Chakraborty AR, Basseville A, et al. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm. 2011; 8: 2021?2031. doi:10.1021/mp200329f