
Inflammatory Mechanisms Contributing to Aortic Expansion and Rupture after Acute Aortic Dissection
Atsushi Anzai*, Motoaki Sano
Department of Cardiology, Keio University School of Medicine, Tokyo, Japan
Abstract
In-hospital outcomes are generally acceptable with the conservative treatment of uncomplicated type B aortic dissection, but some patients present with undesirable complications, such as aortic expansion and rupture. Beyond mechanical and shear forces of blood flow affecting the weakened aortic wall, excessive inflammatory response has been shown to be associated with aortic expansion and adverse clinical outcomes. We have previously demonstrated the underlying mechanisms of catastrophic complications after acute aortic dissection (AAD) in mice. We propose that aortic dissection induces expression of the neutrophil chemoattractants CXCL1 and granulocyte-colony stimulating factor in the aortic tunica adventitia. These local environmental changes recruit neutrophils in combination with alteration of bone marrow milieu where reduced CXCL12 expression enhances neutrophil egress. Interleukin (IL)-6 production in the inflammatory adventitial neutrophils causes vascular inflammation, leading to vascular wall fragility. Targeting CXCR2 or IL-6 mitigates aortic expansion and prevents mice from aortic rupture. Collectively, adventitial neutrophil-mediated inflammation may be a potential therapeutic target to limit lethal complications after AAD.
Acute Aortic Dissection and Inflammation
Acute aortic dissection (AAD) is a life-threatening vascular disease initiated by a tear in the aortic intima1. Subsequent disruption of the medial layer separates the aortic wall into true and false lumens with or without communication. The disease propagates anterogradely or retrogradely, causing a variety of complications depending on the organs and vessel branches involved. The incidence is higher in men than in women and increases with age2. Other common risk factors associated with AAD are hypertension, pre-existing aortic diseases, family history, smoking, chest trauma, and use of intravenous drugs such as cocaine and amphetamines. The Stanford system classifies dissections that involve the ascending aorta as type A, regardless of where the primary intimal tear starts, and all other dissections as type B. Type A AAD is a catastrophic event with high mortality due to the frequency of fatal complications such as aortic rupture, aortic regurgitation, and cardiac tamponade; it usually requires emergency surgical repair as the initial therapeutic option. Type B AAD generally produces more acceptable outcomes without surgery. Spontaneous distal reentry of blood flow into the true lumen and thrombus formation within the false lumen sometimes resolve the disease propagation and help to avoid aneurysm expansion. Although pharmacological treatment targeting high blood pressure and intolerant pain, alongside careful follow up with imaging modalities, improves clinical outcomes in patients with type B AAD, a substantial proportion of medically treated patients still encounter catastrophic events such as aortic expansion and subsequent aortic rupture even after beginning appropriate management3, 4. During the natural history of medically managed type B AAD patients, the fatal complications usually occur within 15 days of presentation5, suggesting the importance of further understanding of the disease pathophysiology especially in the acute phase. What a key mediator that incites AAD progression and causes adverse outcomes? Beyond hemodynamic forces affecting the weakened aortic wall, several clinical studies proposed that excessive inflammation is implicated in disease development and is a potential therapeutic target. Peak C-reactive protein (CRP), for example, is a strong predictor of early and late adverse complications in patients with type B AAD6-9. In one retrospective observation, peak serum CRP concentration during hospitalization was positively correlated with the magnitude of aortic enlargement and oxygenation impairment, resulting in poor in-hospital prognosis for patients with distal AAD6. Imaging analysis also showed that fluorodeoxyglucose uptake, evidenced by positron emission tomography in the dissected aorta, predicted worse long-term clinical outcomes after type B AAD9. These data suggest that systemic activation of the inflammatory cascade following AAD may be critical in the development of catastrophic complications.
Inflammatory Response Following AAD
Experimental approaches are necessary to explore whether enhanced inflammatory response after AAD has a causal effect on disease progression. Previous preclinical studies have focused on the mechanisms for AAD initiation via traditional mouse models, which depend on long-term infusion of angiotensin II (AngII) in ApoE knockout or aged mice10, 11. Prevalence of AAD in such animals is quite low and occurrence of the disease is by chance, suggesting the difficulty of applying such models to investigate what occurs after aortic injury. Kurihara et al. established a novel mouse model in which AAD develops at 100% within 24 hours after starting continuous AngII infusion12. This model relies on a lysyl oxidase inhibitor β-aminopropionitrile monofumarate (BAPN)-induced preconditioning that weakens collagen and elastin crosslinks in the vessel wall in immature mice. While this pre-AAD aortic condition is typically seen in connective tissue diseases, such as Marfan and Ehlers–Danlos syndromes, it also arises in connection to aging and atherosclerosis13, 14. Likewise, serum levels of AngII, the potential trigger cracking the aortic intima in the model, is significantly elevated in human AAD patients compared to healthy controls and non-ruptured aneurysm patients12. This new mouse model therefore recapitulates key features of human aortic dissection and facilitates in vivo investigation of the natural course after disease onset.
We have previously shown that AAD unleashes an inflammatory response that causes disease progression15. In response to AngII, dissection usually starts at the proximal site of the descending thoracic aorta and propagates distally along with aortic expansion. As a consequence, around 70% of the mice eventually die of aortic rupture within 48 hours after the onset of AngII. Histopathological examination accompanied by flow cytometric analysis reveals the spatiotemporal dynamics of inflammatory cell infiltration to the dissected aorta, where neutrophils massively accumulate in the tunica adventitia until 48 hours after the onset of AngII. Since negligible neutrophils are observed in non-dissected aortic adventitia in BAPN-preconditioned mice with AngII, accumulation of those adventitial neutrophils is a result of aortic dissection. Macrophage and T cell counts are also increased in the tunica adventitia of the injured aorta, and neutrophils dominate the leukocyte populations. These temporal and spatial characteristics of the leukocyte accumulation pattern are consistent with previously reported human AAD pathology16. Blood neutrophils increase over time in AAD mice, whereas monocytes and T cells in the blood are not affected. This suggests that the injured aorta recruits neutrophils from elsewhere through circulation, rather than expanding neutrophils locally. Expression of inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α is augmented peaking at 24 hours after AngII onset; this corresponds to the time course of leukocyte infiltration. Adventitial neutrophils are highly proinflammatory as they are the predominant producer of interleukin-6 (IL-6) especially on the edge of the ruptured site, drawing attention to the activated neutrophils as a crucial player for the disease progression and catastrophic complications.
How are these neutrophils recruited to the dissected aorta? In response to AAD, adventitial cells such as myofibroblasts and macrophages express CXCL1 and granulocyte-colony stimulating factor (G-CSF), known as neutrophil-attracting factors17, 18. These changes appear as early as 6 hours after initiation of AngII infusion, prior to neutrophil accumulation. Aortic wall stretching associated with pseudolumen formation promotes the conversion of fibroblasts into smooth muscle actin (SMA+) myofibroblasts and also stimulates F4/80+ CD206+ resident macrophages in the tunica adventitia. Activation of these cells results in strong CXCL1 production, leading to neutrophil recruitment to the injured site. AAD also facilitates neutrophil egress from the bone marrow (BM) to the circulation. BM protein levels of CXCL12, a stromal cell–producing chemokine that retains neutrophils19, 20, are decreased in AAD mice compared to non-dissected animals. This alteration in the BM milieu is attributed in part to enhanced systemic G-CSF levels, and CXCL1 likewise contributes to augmented BM neutrophil release. Although an increased concentration of circulating CXCL1 and G-CSF is observed in the AAD mice, serum CXCL2 concentration was not augmented despite a significant increase in CXCL2 gene expression in the dissected aorta. This suggests distinct functions for CXCL1 and CXCL2 in the post-AAD inflammatory processes similar to other acute inflammatory conditions21. Altogether, those data indicate that the dissected aorta gives the chemotactic cue to recruit neutrophils from the BM to the tunica adventitia.
Potential New Therapy Targeting Adventitial Neutrophils
To determine whether neutrophil mobilization from the BM plays a causative role in lethal aortic rupture in AAD mice, we took advantage of a neutralizing approach for CXCR215. Though anti–CXCR2 neutralizing antibody did not affect AAD incidence and systolic blood pressure 48 hours after the onset of AngII infusion, the number of neutrophils in the blood and aorta was profoundly reduced, and the survival rate was significantly improved in AAD mice receiving the antibody. Both dissection length and cross-sectional diameter evidenced by contrast-enhanced computed tomography at 48 hours with AngII were also significantly decreased by CXCR2 neutralization. In contrast, the anti–G-CSF neutralizing antibody had a minimal effect on survival in AAD mice, although those mice had comparable disease incidence and blood pressure with significantly reduced blood and aortic neutrophil numbers in comparison to control IgG–treated mice. This difference in efficacy between the two antibodies can be accounted by cells that express their receptors (i.e.. CXCR2 or G-CSF receptor (G-CSFR)). In other words, CXCR2 is exclusively expressed by neutrophils, whereas G-CSFR is expressed on not only neutrophils but also other hematopoietic and nonhematopoietic cells. It can be speculated that G-CSF signaling in cells other than the infiltrating neutrophils may play a protective role in the dissected aorta.
The tunica adventitia of the dissected aorta displays high levels of IL-6 in response to AAD. Most IL-6–producing CD45+ leukocytes in the dissected aorta are neutrophils, as evidenced by flow cytometric and histological analyses. Strong expression of IL-6 on neutrophils can be observed at the edge of the rupture site. Importantly, aortic gene expression and serum concentrations of IL-6 were significantly reduced in AAD mice with CXCR2 neutralization compared to control IgG treatment. This suggests that the effect of CXCR2 neutralization relies in part on attenuation of IL-6–mediated adventitial inflammation. Mice deficient in IL-6 showed improved aortic dilatation and survival without affecting AAD incidence, blood pressure, and blood and aortic neutrophil numbers. Collectively, these data revealed the causal effect of neutrophil-derived IL-6 and the potential therapeutic impact of anti-CXCR2 or anti-IL-6 in preventing lethal complications after AAD.
Potential Translation From Bench to Bedside and Open Questions
Compared to patients with thoracic aortic aneurysm (TAA), patients with AAD have increased serum levels of IL-6, as well as CXCL8 and G-CSF, both of which are neutrophil chemoattractants in humans15. Although there is no difference in their serum concentrations at admission between patients with TAA and AAD, they are significantly elevated in response to AAD, peaking 1 to 2 days after admission. Histopathological examinations using human autopsied dissected aortic tissues reveal predominant expression of IL-6 in adventitial neutrophils. Although the sample size was small, these human data suggest a common pathway leading to aortic expansion and subsequent rupture between mice and humans.
The mechanism by which neutrophils produce IL-6 has not yet been explored. Broken aortic cells release their endogenous components known as danger-associated molecular patterns, and neutrophils might be able to produce and secrete IL-6 upon recognition of those proteins, or perhaps other cells sense them and activate neutrophils for cytokine production. Blockade of this pathway might be an alternative therapeutic strategy to limit AAD progression. Determining appropriate timing for anti-CXCR2 drug administration remains a challenge. Treatment prior to disease onset cannot be translated to the clinic, so further preclinical study—potentially with large animals—is necessary to optimize the treatment regimen. Wu et al. demonstrated that macrophage-derived matrix metalloproteinase-9 participates in acute lung injury, one of the frequent inflammatory complications following AAD22. It is a matter of great interest whether neutrophil-derived IL-6 also contributes to the underlying pathogenesis in such a systemic event. Recent clinical advances suggest endovascular repair as an emerging therapeutic strategy for patients with both complicated and uncomplicated aortic dissection2, 23, 24. The effect of excessive inflammation on outcomes for patients who have undergone stent grafting remains unclear. It might be feasible to target neutrophil-mediated post-AAD inflammation in combination with endovascular repair.
Conclusions
Aortic dissection per se induces neutrophil chemoattractant expression in the aortic tunica adventitia, possibly by mechanical injury and stretching followed by pseudolumen formation. Subsequent systemic changes in chemokine-dependent signaling cause neutrophilia and massive neutrophil accumulation in the tunica adventitia of the dissected aorta, leading to aortic expansion and rupture via IL-6 production (summarized in Figure 1). Importantly, the temporal and spatial dynamics of inflammatory cytokine and chemokine elevation, as well as leukocyte accumulation are consistent between rodents and humans. These new insights associated with neutrophil-mediated adventitial inflammation provide us novel therapeutic strategies for patients with AAD.
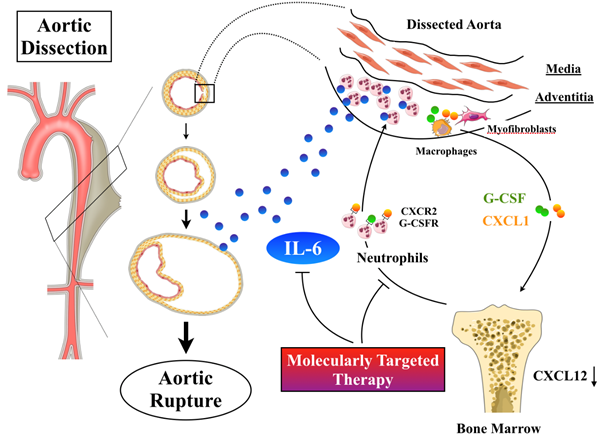
Figure 1. Inflammatory mechanisms contributing to AAD progression. Aortic dissection induces expression of CXCL1 and G-CSF in the tunica adventitia of the aorta, possibly by mechanical injury and stretching followed by pseudolumen formation. These environmental changes recruit neutrophils in combination with alteration of bone marrow where neutrophil egress is enhanced. Interleukin (IL)-6 production in inflammatory adventitial neutrophils causes vascular inflammation by the outside-in mechanism, leading to vascular wall fragility.
Conflict of Interest Statement
We declare no financial conflict of interest.
Source of Funding
This work was supported by JSPS KEKENHI Grant Number 18K08048 (to A. Anzai).
Acknowledgements
The figure was designed in part using Servier Medical Art (http://www.servier.com).
Disclosures
None.
References
- Sheikh AS, Ali K, Mazhar S. Acute aortic syndrome. Circulation. 2013; 128: 1122–1127.
- Erbel R, Aboyans V, Boileau C, et al. ESC Committee for Practice Guidelines. Eur Heart J. 2014; 35: 2873-2926.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. The International Registry Of Acute Aortic Dissection (Irad): new insights into an old disease. JAMA. 2000; 283: 897–903.
- Tsai TT, Trimarchi S, Nienaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg. 2009; 37: 149–159.
- Durham CA, Cambria RP, Wang LJ, et al. The natural history of medically managed acute type B aortic dissection. J Vasc Surg. 2015; 61: 1192–1198.
- Sugano Y, Anzai T, Yoshikawa T, et al. Serum C-reactive protein elevation predicts poor clinical outcome in patients with distal type acute aortic dissection: association with the occurrence of oxygenation impairment. Int J Cardiol. 2005; 102: 39–45.
- Kurabayashi M, Okishige K, Azegami K, et al. Reduction of the PaO2/FiO2 ratio in acute aortic dissection – relationship between the extent of dissection and inflammation. Circ J. 2010; 74: 2066–2073.
- Sakakura K, Kubo N, Ako J, et al. Peak C-reactive protein level predicts long-term outcomes in type B acute aortic dissection. Hypertension. 2010; 55: 422–429.
- Kato K, Nishio A, Kato N, et al. Uptake of 18F-FDG in acute aortic dissection: a determinant of unfavorable outcome. J Nucl Med. 2010; 51: 674–681.
- Saraff K, Babamusta F, Cassis LA, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003; 23: 1621–1626.
- Tieu BC, Lee C, Sun H, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009; 119: 3637–3651.
- Kurihara T, Shimizu-Hirota R, Shimoda M, et al. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012; 126: 3070–3080.
- Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. 1977; 39: 13–20.
- Carlson RG, Lillehei CW, Edwards JE. Cystic medial necrosis of the ascending aorta in relation to age and hypertension. Am J Cardiol. 1970; 25: 411–415.
- Anzai A, Shimoda M, Endo J, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015; 116: 612-623.
- Xu L, Burke A. Acute medial dissection of the ascending aorta: evolution of reactive histologic changes. Am J Surg Pathol. 2013; 37: 1275–1282.
- Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. 2011; 32: 452–460.
- Summers C, Rankin SM, Condliffe AM, et al. Neutrophil kinetics in health and disease. Trends Immunol. 2010; 31: 318–324.
- Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr Opin Hematol. 2007; 14: 3–8.
- Martin C, Burdon PC, Bridger G, et al. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003; 19: 583–593./li>
- Schiwon M, Weisheit C, Franken L, et al. Crosstalk between sentinel and helper macrophages permits neutrophil migration into infected uroepithelium. Cell. 2014; 156: 456–468.
- Wu Z, Ruan Y, Chang J, et al. Angiotensin II is related to the acute aortic dissection complicated with lung injury through mediating the release of MMP9 from macrophages. Am J Transl Res. 2016; 8: 1426-1436.
- Grabenwoger M, Alfonso F, Bachet J, et al. Thoracic Endovascular Aortic Repair (TEVAR) for the treatment of aortic diseases: a position statement from the European Association for Cardio-Thoracic Surgery (EACTS) and the European Society of Cardiology (ESC), in collaboration with the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J 2012; 33: 1558–1563.
- Fattori R, Montgomery D, Lovato L, et al. Survival after endovascular therapy in patients with type B aortic dissection: a report from the International Registry of Acute Aortic Dissection (IRAD). JACC Cardiovasc Interv. 2013; 6: 876–882.