
MHC-1 Mediated Antigen Presentation Machinery as a Key of Tumor Cells Immune Escape Control
Christian B. Auclair* & Annette Ives#
AC BioScience SA, Biopôle, Route de la Corniche 4 – Lysine, 1066 Epalinges, Switzerland
Background
Immune escape is one of the key mechanisms mediating cancer progression and metastatic invasion. This creates significant obstacles to successful cancer treatment, especially those reliant on the immune response such as T cell adoptive immunotherapy and immune checkpoint inhibitors (ICI) therapy. Despite a functioning host immune system, tumor cells often acquire either poorly immunogenic or immunosuppressive phenotypes, which impair immune-dependent resolution.
During initial tumor development, most tumor cells are recognized and efficiently cleared by the immune system. Yet, this creates a selection pressure, whereby the emergence of resistant variants occurs through immunoediting. These variants often lie dormant in patients during the "equilibrium phase" prior to re-emerging with malignant phenotypes during the "escape phase" leading to immune subversion.
Immunoediting is a crucial mechanism in tumor evasion of immune surveillance1. Consequently, nascent malignant cells acquire defects in antigen presentation and lose the surface expression of tumor antigen-derived peptides in the context of Major Histocompatibility Complex (MHC-I). This reduced- or lack of- expression results in impaired ligation with the T cell receptor (TCR) on the surface of CD4 and CD8 T cells leading to defects in immunological synapse formation. Thus, abrogating the global immune response amplitude and quality of T-cell priming, activation, proliferation, differentiation, and cytokine production. The further acquisition of immunosuppressive properties, such as expression of PD-L1 and secretion of suppressive cytokines leads to T-reg expansion and macrophage M2 phenotype.
Impairment of MHC-1 Mediated Antigen Presentation Machinery in Tumor Cells
MHC-I dependent (also known as human leukocyte antigen (HLA-I) immune recognition of tumor antigens is a crucial mechanism through which tumor cells are recognized and cleared by the host. Within tumor cells, a discrete and multi-step process occurs, all of which can be targeted for immune subversion.
Tumor antigens are translocated from the cytosol to the ER by the transporter associated with antigen processing (TAP)2 and then processed into 8-10 amino acid peptides by the immunoproteasome involving proteins for proteasome assembly (PSMB) as well as endoplasmic reticulum aminopeptidases (ERAP). Concurrently, ER-resident chaperones, including calreticulin and ERP573, facilitate the folding of nascent MHC-I molecules. Subsequently, the heavy chain and β2M of the MHC complex binds to TAP, in the presence of Tapasin to stabilize the complex and facilitate the binding of high-affinity peptides into the MHC-I. After TAP dissociation, MHC-I: peptide complexes use Golgi apparatus-dependent vesicular transport to traffic to the cell membrane.
It is well established in both human and experimental tumor models that tumor immune escape is associated with MHC-I downregulation5-6.
MHC-I expression depends on the tumor phenotype and two extreme phenotypes can be identified: a "good" phenotype (rejected tumor phenotype) and a "bad" phenotype (escape tumor phenotype). Tumors with the latter profile are derived from tumor progression following T-cell mediated immunosurveillance escape7. Tumors derived from (HLA-I) positive epithelia can lose totally or partially the surface expression of MHC-I molecules8. The total percentage of various types of HLA-I loss, including complete loss, haplotype loss, or allelic loss, ranges from 65 to 90%, depending on the type of cancer9.
The relevance of the phenotypic classification is provided as a typical example by colorectal cancer (CRC) patients in which 35% of them have an abnormal MHC-I expression. TAP1 downregulation mediates immune escape and is associated with a poor prognosis in CRC patients10. This observation was further supported by the elegant work of Wang et al.11 whereby for most tumor types a clear correlation exists between pan-cancer immune checkpoint inhibitors objective response rates (ORR) and immune tumor status for individual patients. These results were based on tumor immunogenicity scores (TIGS) that combined tumor mutational burden (TMB) with the expression signature of the antigen processing and presenting machinery (APM).
Moving Tumor Cells from an Escape Phenotype to a Rejected Phenotype and Restauration of MHC-I
The escape phenotype is integral for malignancy and is an outcome of tumor cell reprogramming. Genetic and epigenetic processes govern this process and induce global gene expression modulation and alter protein expression. Following "the escape" process, tumor cells not only indicate immunotherapeutic resistance but display a malignant phenotype including abnormal metabolism, escape from growth factor control, stemness and invasive characters12. Hence, the immune-refractory phenotype can be associated with malignant phenotypes and are linked by factors that act as the regulators of malignant transformation and maintenance.
In this context, the challenge is to define strategies that "reprogram" tumor cells towards an elimination phenotype capable of presenting tumor antigens and display minimal immunosuppressive properties. As such, development of a genetic or pharmacologic combination approach comprised of ICI and an immunomodulatory agent is extremely attractive.
It has been recently published13 that cancer cells transfected by the tumor suppressor Fhit (fragile histidine triad) restored MHC-I expression, improved tumor immunogenicity and promoted tumor rejection in immunocompetent mice mediated via CD8+ T cell-mediated immune response development. Fhit is a dinucleoside 5',5"'-P1, P3-triphosphate (Ap3A) hydrolase involved dinucleoside polyphosphate signal transduction. Fhit displays several biological functions, including for genome integrity and the regulation of the gene expression and translation. Fhit transfection of tumor cells induces phenotypic changes resulting in MHC-I mediated antigen presentation and overall immune activation, providing support for the importance of new strategies for tumor phenotype switching from an "escape" to "rejected" status (Figure 1). Along this line, tumor malignancy can be reversed14, and a large set of genes are implicated- and can thus be targeted for cancer treatment. Differential gene-expression profiling has identified, amongst others-SIAH-1, PS1, TSAP6, and, most importantly, translationally controlled tumor protein (TCTP) and TP53. Decreasing TCTP expression and concomitant increased of TP53 expression is key in reprogramming malignant cells, including cancer stem cells15.
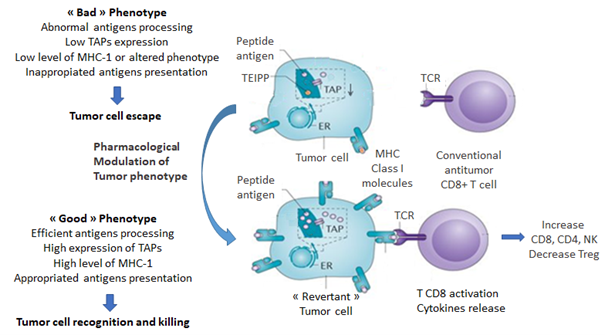
Figure 1: Pharmacological-mediated tumor cell phenotype reversion results in the shift from escape to rejected phenotype. TAP: transporter associated with antigen processing, TEIPP: T-cell epitopes associated with impaired peptide processing, TCR: T Cell Receptor.
In terms of pharmacology, it has been shown that neuromodulating drugs such as sertraline, thioridazine16, and more recently, a Peganum harmala beta-carboline derivative induces tumor reversion17. Revertant cells are characterized by cytoskeletal remodeling18, increased cell-cell adhesion forces and a loss of growth properties in semi-solid supports.
Actin-dependent cytoskeletal remodeling and membrane/vesicle trafficking play multiple critical roles in T and B lymphocytes19,20, especially during immunological synapse formation of their respective T-cell receptors (TCR) and B-cell receptors (BCR) with either MHC-I expressing tumor cells or antigen-presenting cells. Actin dynamics, including F-actin, mediate clustering, segregation, and receptor movement at the synapse interface21,22. Thus, facilitating synapse formation, strength, and duration to influence the development of anti-tumor adaptive immune responses.
As previously mentioned, recent experiments23 demonstrated that a beta-carboline (referred to as ACB1801)-mediated tumor reversion resulted in the rescue of MHC-I antigen presentation in melanoma B16 F-10, a poorly immunogenic murine tumor resistant to anti-PD-1. ACB1801 treatment of B16 F10 tumor cells increased the expression of antigen-specific MHC-I: peptide complexes at the cell, which correlated with increased levels of TAP-1, TAP-2 and tapasin gene expression that all act together to mediate antigen processing. These effects are like those induced by IFN-y, but in contrast to interferon, the drug does not increase PDL-1 expression. In vivo, ACB1801 strongly potentiates the effect of the anti-PD-1 with reduced tumor growth and improved survival.
It should be emphasized i) that the efficacy of cancer immunotherapy is primarily dependent on MHC-I mediated antigen presentation in tumor cells, ii) that the rescue of antigen presentation can be achieved through a non-cytotoxic reversion of the malignant phenotype and iii) that the phenotype reversion can be induced using small chemical entities in experimental models. These small chemical entities could further be used during adoptive cell therapies to facilitate ex vivo selection, and expansion of tumor-killing CD8+ T-cells, and improve MHC-I: tumor peptide antigen-dependent tumor targeting if patients are administered with the compounds at the time of transfer.
Altogether, these possibilities pave the way for new strategies to improve the response rate and efficacy of ICI therapy.
References
- O'Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 16, 151–167 (2019). https://doi.org/ 10.1038/s41571-018-0142-8
- Abele R, Tampé R. Function of the transport complex TAP in cellular immune recognition. Biochim Biophys Acta. 1999 Dec 6;1461(2):405-19. doi: 10.1016/s0005-2736(99)00171-6. PMID: 10581370.
- Antoniou AN, Ford S, Alphey M, et al. (2002) The oxidoreductase ERP57 efficiently reduces partially folded in preference to fully folded MHC class I molecules. EMBO J. 21, 2655–2663.
- Spiliotis ET, Osorio M, Zuniga M, et al. Selective Export of MHC Class I Molecules from the ER after Their Dissociation from TAP Immunity March 200113(6): 841-51 DOI: 10.1016/S1074-7613(00)00081-9
- Seliger B, Maeurer M, Ferrone S. (2000) Antigen-processing machinery breakdown and tumor growth. Immunol Today 21: 455–464
- Garrido F, Aptsiauri N, Doorduijn EM, et al. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016; 39: 44-51. doi: 10.1016/j.coi.2015.12.007
- Garrido F, Algarra I. MHC antigens and tumor escape from immune surveillance. Adv Cancer Res. 2001; 83:117-58. doi: 10.1016/s0065-230x(01)83005-0. PMID: 11665717.
- Garrido F, Ruiz-Cabello F, Cabrera T, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today. 1997 Feb; 18(2):89-95. doi: 10.1016/s0167-5699(96)10075-x. PMID: 9057360.
- Menon A, Morreau H, Tollenaar R, et al. Down-Regulation of HLA-A Expression Correlates with a Better Prognosis in Colorectal Cancer Patients. Lab Invest 82, 1725–1733 (2002). https://doi.org/10.1097/01.LAB.0000043124.75633.ED
- Ling A, Löfgren-Burström A, Larsson P, et al. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology. 2017; 6(11):1–10. https://doi.org/ 10.1080/2162402X.2017.1356143.
- Wang S, He Z, Wang X, et al. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Cancer Biology, eLife 2019; 8: e49020 doi: 10.7554/eLife.49020
- Oh SJ, Lee J, Kim Y, et al. Far Beyond Cancer Immunotherapy: Reversion of Multi-Malignant Phenotypes of Immunotherapeutic-Resistant Cancer by Targeting the NANOG Signaling Axis. Immune Netw. 2020 Feb; 20(1): e7
- Pulido M, Chamorro V, Romero I, et al. Restoration of MHC-I on Tumor Cells by Fhit Transfection Promotes Immune Rejection and Acts as an Individualized Immunotherapeutic Vaccine. Cancers (Basel). 2020 Jun 12; 12(6): 1563. doi: 10.3390/cancers12061563. PMID: 32545680; PMCID: PMC7352176.
- Powers S, Pollack R. Inducing stable reversion to achieve cancer control. Nat Rev Cancer 16, 266–270 (2016). https://doi.org/10.1038/nrc.2016.12
- Amson R, Karp JE, Telerman A. Lessons from tumor reversion for cancer treatment. Curr Opin Oncol. 2013 Jan; 25(1): 59-65. doi: 10.1097/CCO.0b013e32835b7d21.
- Tuynder M, Fiucci G, Prieur S, et al. Translationally controlled tumor protein is a target of tumor reversion. Proc Natl Acad Sci U S A. 2004 Oct 26;101(43):15364-9. doi: 10.1073/pnas.0406776101. Epub 2004 Oct 15. PMID: 15489264; PMCID: PMC523462.
- Le Moigne R, Subra F, Karam M, et al. The β-Carboline Harmine Induces Actin Dynamic Remodeling and Abrogates the Malignant Phenotype in Tumorigenic Cells. Cells. 2020 May 8; 9(5): 1168. doi: 10.3390/cells9051168. PMID: 32397195; PMCID: PMC7290983.
- Weber K, Lazarides E, Goldman RD, et al. Localization and distribution of actin fibers in normal transformed and revertant cells. Cold Spring Harb Symp Quant Biol 1975, 39 Pt 1:363-369.
- Chichili GR, Westmuckett AD, Rodgers W. T cell signal regulation by the actin cytoskeleton. J. Biol. Chem. 285, 14737–14746 (2010).
- Colin A, Bonnemay L, Gayrard C, et al. Triggering signaling pathways using F-actin self-organization. Sci Rep 6, 34657 (2016). https://doi.org/10.1038/srep34657
- Kumari S, Curado S, Mayya V, et al. T cell antigen receptor activation and actin cytoskeleton remodeling, Biochimica et Biophysica Acta - Biomembranes. 2014; 1838, 2, 546-556
- Brown BK and Song W, The Actin Cytoskeleton is Required for the Trafficking of the B Cell Antigen Receptor to the Late Endosomes. Traffic, 2001; 414-427.
- Schläpfer A, Auclair C, Janji B, et al. The 'Holy Grail' in Immuno-Oncology: AC BioScience SA is Aiming to Potentiate Anti-PD-1 Therapy Efficacy through Tumor Cell Conditioning Strategy. Chimia (Aarau). 2020 Oct 28; 74(10): 771-775. doi: 10.2533/chimia.2020.771. PMID: 33115558.