
Pathophysiological Mechanisms Leading to Low Platelet Count in Immune Thrombocytopenia
Paola Roxana Lev1,2, Nora Paula Goette1, Rosana Fernanda Marta1,2*
1Institute of Medical Research A. Lanari, School of Medicine, University of Buenos Aires, Buenos Aires, Argentina
2Department of Hematology Research, Institute of Medical Research (IDIM), National Scientific and Technical Research Council (CONICET), University of Buenos Aires, Buenos Aires, Argentina
Abstract
Primary immune thrombocytopenia (ITP) is an autoimmune disorder characterized by the decrease in peripheral blood platelet count below 100 x 109/L, and an increased bleeding risk when thrombocytopenia drops below 30 x 109/L. The mechanisms leading to ITP in adults, although not completely elucidated, involves an imbalance between effector and regulatory cells that results in a breakdown of the immune tolerance. Autoantibodies are considered the main responsible for thrombocytopenia, although direct T-cell cytotoxic effect and lysis by Complement attachment and activation could also contribute to platelet elimination from circulation. In addition to increased peripheral clearance, abnormalities in platelet production also favors platelet count reduction. This review is intended to describe some specific knowledge about peripheral and bone marrow mechanisms leading to thrombocytopenia in adult ITP.
Introduction
Primary immune thrombocytopenia (ITP) is an acquired autoimmune disorder characterized by chronic isolated low platelet count (<100 x 109/L)1, triggered by a loss of the immunological system equilibrium that tips the balance toward autoimmunity. Although in the last years the pathophysiology of ITP has been intensively studied, this field still remains incompletely understood2,3. Another significant feature of ITP is the heterogeneity observed among patients, concerning not only their clinical profile but also their response to treatment, with a small proportion of patients classified as refractory4. This heterogeneity is likely due to the participation of multiple pathophysiological contributing mechanisms, affecting individual patients in different degrees.
The presence of autoantibodies targeting platelet surface glycoprotein(s) (GP) has been demonstrated in patients with ITP with percentages of patients with positive autoantibodies varying according to different case series5-7. Most of these antibodies are IgG, but IgM and IgA can also be detected, usually in association with IgG8. Main autoantibody targets are GPIIbIIIa (integrin αIIbβ3), GPIbIX and GPIaIIa complex, but, autoantibodies against other GPs (i.e. the vitronectin receptor, integrin αvβ3)9, against thrombopoietin and/or its receptor c-Mpl10-12 were also detected in some patients. Therefore, humoral immunity has been classically considered the main cause of thrombocytopenia, although other mechanisms such as direct T-cell cytotoxic effect could contribute to platelet elimination as well13,14. In addition, Complement activation/fixation capacity is enhanced in plasma samples from ITP15,17. Indeed, Complement-mediated platelet destruction has been pointed as another mechanism of platelet elimination, mainly triggered by anti-platelet autoantibodies, but also observed in some ITP patients with no-detectable autoantibodies18.
This review summarizes mechanisms leading to thrombocytopenia in ITP with special focus on peripheral platelet destruction and impaired platelet production.
Increased circulating platelet clearance
The first and most commonly accepted mechanism of platelet clearance in ITP is the binding of anti-platelet autoantibodies to their antigenic glycoprotein targets that leads to platelet elimination by monocytes/macrophages of the reticuloendothelial system through FcγRIIA and FcγRIIIA-mediated recognition, mainly in the spleen and liver19. However, additional pathological mechanisms including platelet apoptosis and loss of sialic acid from platelet membrane glycoproteins were described in recent years, that also contribute to platelet clearance from circulation.
Increased platelet apoptosis in ITP
Several studies have demonstrated the contribution of platelet apoptosis to ITP pathogenesis. Winkler and col20 showed evidence of platelet apoptosis, including caspase 3, 8 and 9 activation, in children with acute ITP, which was ameliorated by intravenous immunoglobulin infusion. Concerning adult chronic ITP, increased platelet phosphatidylserine (PS) exposure, as a marker of platelet apoptosis was shown by Catani21 and Alvarez Roman22.
In 2016, our group described increased mitochondrial membrane depolarization, active caspase 3 and membrane exposure of PS in platelets from ITP, as markers of apoptosis, while basal platelet activation assessed by PAC-1 binding and P-selectin externalization was ruled out as another possible cause of increased PS expression23. Later, Deng and col24 confirmed these results and showed higher expression levels of the proapoptotic molecules Bak and Bax, while the antiapoptotic factor Bcl-xL was decreased. Moreover, the same group demonstrated dysregulation of certain microRNAs related to apoptosis in platelets from ITP25.
Cells expressing PS on their membrane are known to be cleared through scavenger receptors in macrophages, thus, apoptotic platelets would also be eliminated from circulation by this mechanism. In our cohort of ITP, the inverse relationship found between apoptotic variables and peripheral platelet counts, further reinforces the relevance of platelet apoptosis in the development of thrombocytopenia in this disorder. Interestingly, platelet apoptosis was observed in all ITP patients carrying auto-antibodies against the glycoproteins GPIIbIIIa and GPIbIX, but was absent in those patients expressing anti-GPIaIIa autoantibodies, suggesting a causal role for these specific antibodies in triggering this phenomenon.
In this study, PS externalization was not noticed in normal platelets incubated with ITP plasmas but it could be observed by adding normal autologous CD3+ lymphocytes to this system, suggesting an antibody-dependent cytotoxic effect. In the same system, anti-platelet antibodies-depleted ITP plasma had no effect on normal platelet apoptosis, while the IgG fraction of ITP plasma retained the apoptotic effect observed when whole plasma was used, confirming the involvement of auto-antibodies in the induction of apoptosis.
Interestingly, recent work described that autophagy is decreased in platelets from ITP26 and suggested this reduction could be linked to increased platelet apoptosis. Further studies are warranted to elucidate the interplay between autoantibodies, autophagy and apoptosis in this disorder.
Increased loss of sialic acid from platelet glycoproteins in ITP
In 2015, Li and col.27 described for the first time that plasma from ITP patients containing anti-GPIbIX autoantibodies induced loss of sialic acid capping carbohydrates from platelet glycoproteins. This mechanism is triggered by the ability of these autoantibodies to induce platelet activation, which promote neuraminidase externalization, the ultimate responsible for sialic acid cleavage from membrane glycoproteins. Platelets bearing asialoglycoproteins on their membrane are clarified in the liver by cooperation between macrophage galactose lectin (MGL) and the C-type lectin receptor CLEC4F, both expressed on Kupffer cells, and Ashwell-Morell receptor (AMR) in hepatocytes, as demonstrated by studies performed in mice28,29. In addition, activation of the JAK2/STAT3 pathway downstream AMR after asialoglycoprotein binding, has been pointed as the mechanism triggering synthesis of thrombopoietin (Tpo), the main growth factor of the megakaryocytic lineage30.
In a recent study, our group investigated several possible causes of thrombocytopenia in a cohort of ITP patients, including platelet desialylation. Results showed that loss of sialic acid from normal platelet glycoproteins occurs not only in the presence of anti-GPIbIX but also anti-GPIIbIIIa ITP autoantibodies, as assessed by the increase in Ricinus communis agglutinin I (RCA-I), recognizing galactosyl (β-1,4) N-acetylglucosamine (Galβ4GlcNAc) and Peanut agglutinin (PNA) which recognizes galactosyl (β-1,3) N-acetylgalactosamine (Galβ3GalNAc), both exposed after sialic acid loss. In addition, desialylation could be concomitantly observed with platelet apoptosis, although this was not a mandatory association31. Recently, Manzano and col reported increased apoptosis and loss of sialic acid in platelets from non-responsive ITP patients32. Interestingly, although a direct correlation was found between caspase activation and desialylation in the whole population included in the study (ITP patients who did not need treatment for at least six months, ITP patients responding to agonists of thrombopoietin receptors, and non-responders), levels of caspase activity between non-responders and thrombopoietin receptor agonists-responders were not different, supporting the notion that apoptosis and desialylation are not necessarily related. In addition, decreased platelet surface glycans in non-responders ITP patients was accompanied by a decrease in T regulatory lymphocyte population suggesting a link between this platelet abnormality and immune imbalance.
The observation that anti-GPIIbIIIa autoantibodies can also induce loss of sialic acid from platelet glycoproteins was recently confirmed by Marini and col33. By blocking the FcγRIIA signaling, authors demonstrated that sialic acid loss is at least partially due to crosslinking of anti-GPIIbIIIa autoantibodies to FcγRIIA, which is responsible for the release of sialidase from the intracellular compartments of platelets.
Concerning circulating Tpo levels in ITP, it was suggested that they were lower than expected for the degree of thrombocytopenia34. In the late ´90s, when Tpo synthesis by the liver was supposed to be constant, the cumulative mass of megakaryocytes and platelets were considered as the main determinant of Tpo levels35,36. Nowadays, assuming that binding of desialylated platelets to hepatic AMR is, at least in part, responsible for Tpo synthesis, differences in platelet desialylation levels among patients with ITP and the fact that some of them do not display increased platelet desialylation, could account for variations in Tpo levels among patients population. The influence of desialylation in modulating circulating Tpo levels in ITP should be tested by comparing these variables in a large ITP cohort. In addition, other factors such as platelet GPIbα were described to regulate hepatic Tpo homeostasis37. In this regard, ITP autoantibodies targeting this glycoprotein could interfere with this mechanism. Therefore, further investigations are needed to fully understand the regulation of Tpo synthesis in ITP as well as in physiologic conditions.
Impaired platelet production
Platelet production is the final step of a complex series of events involving two well defined processes: megakaryopoiesis and thrombopoiesis. Megakaryopoiesis initiates when undifferentiated CD34+ hematopoietic progenitor commits to the megakaryocytic lineage and ends when megakaryocytes reach their full maturational state. Next, the process identified as thrombopoiesis involves cytoplasmic megakaryocytic reorganization, proplatelet extension and branching and, after these proplatelets go through the endothelial bone marrow barrier, platelets are released from their tips into the bloodstream.
Megakaryopoiesis in ITP
Early independent works from Chang38 and McMillan39 studied the effect of the addition of 10% of ITP plasma to the culture medium of normal hematopoietic progenitors induced to differentiate into megakaryocytes. Their results showed that plasma from ITP patients impaired megakaryocyte development, pointing to autoantibodies directed to GPIbIX and GPIIbIIIa as responsible for this effect. Unlike these previous studies that used mononuclear cells39 or 20-30% pure CD34+ progenitors38 as source of normal hematopoietic precursors, we reproduced these experiments using at least 93% pure CD34+ hematopoietic progenitors isolated from normal human cord blood. In these conditions, megakaryopoiesis was not inhibited by ITP samples. Moreover, the expression of CD42b, a marker of megakaryocyte maturation, was increased in the presence of 45% of the ITP plasmas, while megakaryocyte size and ploidy were normal31. These results are in line with the known concept that the number of megakaryocytes in bone marrow from ITP patients is normal or increased40,41.
Besides, although it was described that circulating levels of thrombopoietin are low for the degree of thrombocytopenia in ITP, as mentioned before, they seem sufficient to maintain a proper megakaryocyte population in bone marrow.
Participation of apoptosis during megakaryocyte and platelet production
Reports describing apoptosis in the megakaryocytic lineage performed on bone marrow samples from ITP patients are controversial. Uçar and col42 found no differences in the number of megakaryocytes and their apoptotic status between ITP and control children; Houwerzijl et al43 found ultrastructural abnormalities compatible with apoptosis in adult ITP, and a recent study described a decrease of megakaryocyte apoptosis in bone marrow biopsies of ITP subjects compared to healthy controls44. Although these discrepant results are intriguing and would encourage researchers to continue the investigations, bone marrow samples form ITP are not easily available because bone marrow examination is not generally needed to diagnose ITP.
Beyond megakaryopoiesis, data about the participation of apoptotic events during normal physiologic proplatelet formation and platelet production has also been controversial. While several studies support that localized apoptotic events are needed for mature megakaryocytes to rearrange their cytoplasmic architecture and proceed with proplatelet extension and platelet release, more recent information hold the notion that apoptosis must be restrained during proplatelet formation, and only when platelets have been released, the remaining naked nuclei undergoes apoptosis (reviewed by McArthur45).
Similarly, investigations evaluating the effect of ITP plasma on mature megakaryocyte apoptosis have led to discordant results. Yang et al46 described lower expression of tumor necrosis factor-related apoptosis-inducing ligand, higher expression of Bcl-xL and lower platelet release in normal megakaryocytes incubated with ITP plasma, pointing to decreased apoptosis as a contributing factor to reduced platelet production. In our hands, normal mature megakaryocytes incubated for 48 hours with 10% ITP plasma did not suffer caspase 3-7 activation, and the number of megakaryocytes displaying nuclear picnosis was slightly increased. However, proplatelet formation was significantly decreased as will be discussed later47. Then, although the number of megakaryocytes in bone marrow from ITP is normal or elevated, abnormalities in the apoptotic pathways of mature megakaryocytes could not be ruled out as possible factors leading to a lower number of megakaryocytes capable of producing proplatelets. Indeed, further work is necessary to clarify this topic.
Altered megakaryocyte functionality and impaired thrombopoiesis
Other important players participating in the proper functionality of megakaryocytes are extracellular matrix proteins present in the bone marrow milieu. Megakaryocyte interaction with different matrix proteins play specific roles in megakaryocytic physiology. Two of these essential molecules present in the vascular niche are fibrinogen and von Willebrand factor (vWf). Main receptors for these ligands are GPIIbIIIa, which binds both proteins, and GPIbIX, that recognizes vWf. In addition to their key role in platelet function, GPIIbIIIa and GPIbIXV complexes are involved in megakaryocyte development and platelet production. Proof of their participation in these events are the deep abnormalities observed in macrothrombocytopenia due to mutations leading to partial activation of GPIIbIIIa48, and mutations in GPIbIXV, responsible for Bernard-Soulier syndrome49.
Grodzielski and col50 demonstrated that plasma and specifically the IgG fraction from ITP patients interfere with adhesion and spreading of megakaryocytes. This interference is specific for each receptor-ligand pair depending on the type of autoantibody present. Anti-GPIIbIIIa autoantibodies inhibit megakaryocytic interaction with fibrinogen, reducing adhesion, spreading and signal transduction downstream this receptor, as demonstrated by decreased β3 subunit phosphorylation. Abnormal glycoprotein function was also observed by impaired PAC-1 binding to megakaryocytes incubated with ITP plasmas bearing these autoantibodies47. Similarly, anti-GPIbIX autoantibodies impair megakaryocyte adhesion and spreading to vWf, and preclude Src phosphorylation downstream this receptor.
The main raison detre of megakaryocytes is platelet production, and, as a consequence of autoantibody binding to GPIIbIIIa and/or GPIbIX complexes, proplatelet formation is inhibited47,51. The participation of autoantibodies in abnormal thrombopoiesis was proved by reproducing the inhibitory effect of ITP plasma using purified IgG, and by its reversion when ITP plasma was immunodepleted from autoantibodies by incubation with normal platelets.
In addition to the decrease in the number of megakaryocytes producing proplatelets, morphological changes were observed in the architecture of proplatelets generated in the presence of ITP plasma. Most of the patient´s samples induced wider and shorter proplatelets, bearing lower numbers of bifurcation points and tips, and overall, less complex than those observed when megakaryocytes were cultured with the addition of normal plasma47. Considering that megakaryocytes must elongate their proplatelets through the bone marrow endothelial barrier in order to release platelets into the bloodstream, these morphological proplatelet features could hinder thrombopoiesis. Altogether, these results could imply that, in addition to the lower number of megakaryocytes capable of producing proplatelets in ITP, each proplatelet-bearing megakaryocyte would be unable to produce a proper number of platelets.
The osteoblastic niche of bone marrow is a type I collagen-enriched environment. Megakaryocyte binding to collagen through GPIaIIa complex inhibits proplatelet production52, precluding premature platelet release within this compartment. Our in vitro experiments demonstrated that plasma and purified IgG fractions from ITP patients containing anti-GPIaIIa autoantibodies, induced a decrease in megakaryocyte adhesion and spreading on type I collagen along with lower MLC2 phosphorylation downstream GPIaIIa50. In agreement with the fact that MLC2 phosphorylation is a key downstream effector mediating GPIaIIa-induced negative regulation of thrombopoiesis53, these autoantibodies interfered with normal inhibition of proplatelet formation on type I collagen, allowing megakaryocytes to extend proplatelets even on a type I collagen-coated surface47. These findings suggest that the presence of anti-GPIaIIa autoantibodies could lead to premature proplatelet production in the osteoblastic niche, hindering platelet release within the bloodstream.
Along with these abnormalities, Zeng and col.54 added autoantibodies targeting the vitronectin receptor, to the list of factors responsible for the interference between megakaryocyte adhesion to the vascular niche, and potentially contributing to impair platelet production in ITP.
In addition to platelets, megakaryocyte glycoproteins are also target for autoantibody desialylation. Unlike what was observed in platelets, loss of sialic acid from normal megakaryocytes was only found in the presence of ITP plasma containing anti-GPIbIX autoantibodies31. Marini and col.33 demonstrated that desialylation induced by ITP autoantibodies (either anti GPIIbIIIa or GPIbIX) inhibits megakaryocyte adhesion to the bone marrow extracellular matrix proteins fibrinogen and vWf, leading to limited cell differentiation and reduced ability to extend proplatelets. Thus, besides increasing platelet clearance, desialylation has also been established as a mechanism of impaired platelet production.
Overall, these studies demonstrate that ITP autoantibodies mainly affect specific and different intracellular pathways, depending on their target proteins, interfering with normal megakaryocytic functions within the bone marrow and leading to decreased platelet production.
Conclusions
The growing amount of knowledge concerning pathophysiological abnormalities in ITP and results that evaluated several of these mechanisms in individual patients, show that mechanisms of thrombocytopenia are multifactorial, and frequently involve concomitant defects in platelet production and clearance. One of the main conclusions of our work concerning platelet production is that ITP autoantibodies are more harmful on inhibiting thrombopoiesis than affecting megakaryopoiesis. Regarding platelet clearance, apart from the classical Fc-dependent mechanism, platelet apoptosis and desialylation are additional and independent processes contributing to lower platelet count (Figure 1). These findings provide a rational for the use of combination therapy, including drugs with different mechanisms of action, in patients with highly refractory disease.
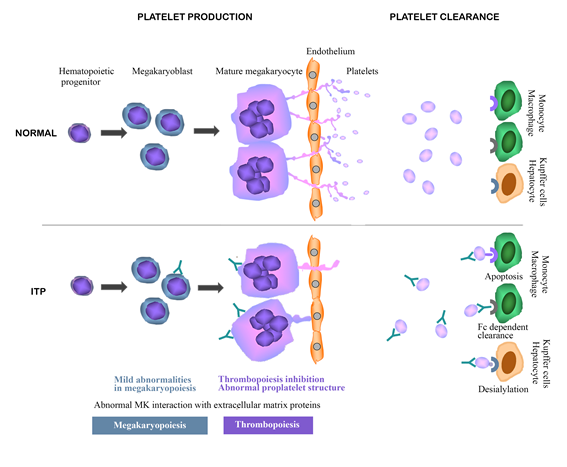
Legend to figure 1: Mechanisms leading to low platelet count in Immune Thrombocytopenia.
This graphical abstract includes mechanisms published in references 19, 24, 37 and 40. Impaired platelet production is likely due to abnormal megakaryocytic function, including altered interaction with extracellular matrix proteins from bone marrow and thrombopoiesis (reduced number of proplatelet-producing megakaryocytes and abnormal proplatelet structure), induced by autoantibody binding to megakaryocytic glycoproteins. Although autoantibody-induced abnormalities were also observed during megakaryopoiesis, they seem to contribute in lesser extent to the reduction in platelet production. Enhanced peripheral platelet clearance could be related to monocyte/macrophage phagocytosis induced by binding of platelet-autoantibody complex to Fcγ receptors IIA and IIIA and phosphatidylserine expressed on apoptotic platelets that binds to scavenger receptors. In addition, desialylated platelets could be clarified by cooperation between Kupffer cells and hepatocytes in the liver.
References
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood. 2009; 113: 2386–2393.
- Li J, Sullivan JA, Ni H. Pathophysiology of immune thrombocytopenia. Curr Opin Hematol. 2018; 25: 373-381.
- Zufferey A, Kapur R, Semple JW. Pathogenesis and therapeutic mechanisms in Immune Thrombocytopenia (ITP). J Clin Med. 2017; 6: 16.
- Witkowski M, Witkowska M, Robak T. Autoimmune thrombocytopenia: Current treatment options in adults with a focus on novel drugs. Eur J Haematol. 2019; 103: 531-541.
- Davoren A, Bussel J, Curtis BR, et al. Prospective evaluation of a new platelet glycoprotein (GP)-specific assay (PakAuto) in the diagnosis of autoimmune thrombocytopenia (AITP). Am J Hematol. 2005; 78: 193-197.
- Stockelberg D, Hou M, Jacobsson S, et al. Detection of Platelet antibodies in chronic idiopathic thrombocytopenic purpura (ITP). A comparative study using flow cytometry, a whole platelet ELISA, and an antigen capture ELISA. Eur J Haematol. 1996; 56: 72-77.
- McMillan R. Antiplatelet Antibodies in Chronic Immune Thrombocytopenia and Their Role in Platelet Destruction and Defective Platelet Production. Hematol Oncol Clin N Am. 2009; 23: 1163–1175.
- He R, Reid DM, Jones CE, et al. Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood. 1994; 83: 1024–1032.
- Kosugi S, Tomiyama Y, Honda S, et al. Anti-alpha(v) beta(3) antibodies in chronic immune thrombocytopenic purpura. Thromb Haemost. 2001; 85: 36–41.
- Kuwana M, Okazaki Y, Kajihara M, et al. Autoantibody to c-Mpl (thrombopoietin receptor) in systemic lupus erythematosus: relationship to thrombocytopenia with megakaryocytic hypoplasia. Arthritis Rheum. 2002; 46: 2148–2159.
- Jing FM, Zhang XL, Meng FL, et al. Anti-c-Mpl antibodies in immune thrombocytopenia suppress thrombopoiesis and decrease response to rhTPO. Thromb Res. 2018; 170: 200-206.
- Nazy I, Kelton JG, Moore JC, et al. Autoantibodies to thrombopoietin and the thrombopoietin receptor in patients with immune thrombocytopenia. Br J Haematol. 2018; 181: 234–241.
- Olsson B, Andersson PO, Jernas M, et al. T-cell mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med. 2003; 9: 1123-1124.
- Zhao C, Li X, Zhang F, et al. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica. 2008; 93: 1428-1430.
- Tsubakio T, Tani P, Curd JG, et al. Complement activation in vitro by antiplatelet antibodies in chronic immune thrombocytopenic purpura. Br J Haematol. 1986; 63: 293–300.
- Peerschke EIB, Andemariam B, Yin W, et al. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with Immune Thrombocytopenic Purpura. Br J Haematol. 2010; 148: 638–645.
- Ge J, Wang L, Li J, et al. Increase of C3a is associated with hemorrhagic propensity in patients with Immune Thrombocytopenia. Clin Lab. 2017; 63: 765-771.
- Najaoui A, Bakchoul T, Stoy J, et al. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur J Haematol. 1012; 88: 167–174.
- McMillan R. The pathogenesis of chronic immune thrombocytopenic purpura. Semin Hematol. 2007; 44: S3–S11.
- Winkler J, Kroiss S, Rand M, et al. Platelet apoptosis in paediatric immune thrombocytopenia is ameliorated by intravenous immunoglobulin. Br J Haematol. 2012; 156: 508–515.
- Catani L, Fagioli ME, Tazzari PL, et al. Dendritic cells of immune thrombocytopenic purpura (ITP) show increased capacity to present apoptotic platelets to T lymphocytes. Exp Hematol. 2006; 34: 879–887.
- Alvarez Roman MT, Fernandez Bello I, Arias-Salgado EG, et al. Effects of thrombopoietin receptor agonists on procoagulant state in patients with immune thrombocytopenia. J Thromb Haemost. 2014; 112: 65– 72.
- Goette NP, Glembotsky AC, Lev PR, et al. Platelet apoptosis in adult immune thrombocytopenia: insights into the mechanisms of damage triggered by auto-antibodies. PLoS One. 2016; 11(8): e0160563.
- Deng G, Yu S, Li Q, et al. Investigation of platelet apoptosis in adult patients with chronic immune thrombocytopenia. Hematology. 2017; 22: 155-161.
- Deng G, Yu S, He Y, et al. MicroRNA profiling of platelets from immune thrombocytopenia and target gene prediction. Mol Med Rep. 2017; 16: 2835-2843.
- Wang Ch, Ma S, Bi S, et al. Enhancing autophagy protects platelets in immune thrombocytopenia patients. Ann Transl Med. 2019; 7: 134.
- Li J, van der Wal DE, Zhu G, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Comm. 2015; 6: 7737.
- Li Y, Fu J, Ling Y, et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells PNAS. 2017; 114: 8360-8365.
- Deppermann C, Kratofil RM, Peiseler M, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020. doi.org/10.1084/jem.20190723
- Grozovsky R, Begonja AJ, Liu K, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med. 2015; 21: 47–54.
- Grodzielski M, Goette NP, Glembotsky AC, et al. Multiple concomitant mechanisms contribute to low platelet count in patients with immune thrombocytopenia. Sci Rep. 2019; 18: 9:2208.
- Monzon Manzano E, Alvarez Roman MT, Sanz RJ, et al. Platelet and immune characteristics of immune thrombocytopaenia patients non-responsive to therapy reveal severe immune dysregulation. Br J Haematol. 2020; 189: 943-953. Comment by Kapur R. Analysing therapeutic responses in Immune Thrombocytopaenia: shifting the focus towards immune characteristic. Br J Haematol. 2020; 189: 811-812.
- Marini I, Zlamal J, Faul C, et al. Autoantibody-mediated desialylation impairs human thrombopoiesis and platelet life span. Haematologica. 2019; pii: haematol.2019.236117.
- Kosugi S, Kurata Y, Tomiyama Y, et al. Circulating Thrombopoietin Level in Chronic Immune Thrombocytopenic Purpura. Br J Haematol. 1996; 93: 704-6.
- Emmons RV, Reid DM, Cohen RL, et al. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction, Blood. 1996; 87: 4068–4071.
- Makar RS, Zhukov OS, Sahud MA, et al. Thrombopoietin levels in patients with disorders of platelet production: diagnostic potential and utility in predicting response to TPO receptor agonists. Am. J. Hematol. 2013; 88: 1041–1044.
- Xu M, Li J, Dias Neves MA, et al. GPIba is required for platelet-mediated hepatic thrombopoietin generation. Blood. 2018; 132: 622-634.
- Chang M, Nakagawa PA, Williams SA, et al. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood. 2003; 102: 887-895.
- McMillan R, Wang L, Tomer A, et al. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood. 2004; 103: 1364–9.
- Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood. 1946; 1: 27–50.
- Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost. 2002; 8: 73-6.
- Ucar C, Oren H, Irken G, et al. Investigation of megakaryocyte apoptosis in children with acute and chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 2003; 70: 347–352.
- Houwerzijl EJ, Blom NR, van der Want JJL, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura Blood. 2004; 103: 500-506.
- Vrbensky JR, Nazy I, Toltl LJ, et al. Megakaryocyte apoptosis in immune thrombocytopenia. Platelets. 2018; 29(7): 729-732.
- McArthur K, Chappaz S, Kile BT. Apoptosis in megakaryocytes and platelets: life and death of a lineage. Blood. 201; 131: 605-610.
- Yang L, Wang L, Zhao C, et al. Contributions of TRAIL-mediated megakaryocyte apoptosis to impaired megakaryocyte and platelet production in immune thrombocytopenia. Blood. 2010; 116: 4307–4316.
- Lev PR, Grodzielski M, Goette NP, et al. Impaired proplatelet formation in immune thrombocytopenia: a novel mechanism contributing to decreased platelet count. Br J Haematol. 2014; 165: 854-64.
- Kunishima S, Kashiwagi H, Otsu M, et al. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the αIIbβ3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood. 2011; 117: 5479-5484.
- Strassel C, Eckly A, Leon C, et al. Intrinsic impaired proplatelet formation and microtubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica. 2009; 94: 800-810.
- Grodzielski M, Di Buduo CA, Goette NP, et al. Autoantibodies in immune thrombocytopenia affect the physiological interaction between megakaryocytes and bone marrow extracellular matrix proteins. Br J Haematol. 2018; 183: 319-323.
- Iraqi M, Perdomo J, Yan F, et al. Immune thrombocytopenia: antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica. 2015; 100: 623–32.
- Sabri S, Jandrot-Perrus M, Bertoglio J, et al. Differential regulation of actin stress fiber assembly and proplatelets formation by alpha2beta1 integrin and GPVI in human megakaryocytes. Blood. 2004; 104: 3117–3125.
- Chang Y, Auradee F, Larbret F, et al. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007; 109: 4229-4236.
- Zeng DF, Chen F, Wang S, et al. Autoantibody against integrin αvβ3 contributes to thrombocytopenia by blocking the migration and adhesion of megakaryocytes. J Thromb Haemost. 2018; 16: 1843-1856.