
Sneaking ligands inhibiting NF-kappaB activation selectively in endothelial cells improve experimental arthritides
Bettina Sehnert1*, Harald Burkhardt2, Stefan Dübel3, Reinhard E. Voll1
Abstract
The activation of intracellular signaling pathways such as the classical nuclear factor kappaB (NF-kappaB) pathway is related to the pathogenesis of several inflammatory autoimmune diseases including rheumatoid arthritis (RA). To clarify the role of disease-relevant cell-types and signaling molecules in vitro or in vivo, it is necessary to target them selectively without disturbing the homeostasis of the immune system. We developed sneaking ligand fusion proteins (SLFPs) for cell-type specific modulation of signaling pathways. We designed the first SLFPs to inhibit the activation of NF-kappaB, a key regulator of inflammation, solely in the activated endothelium. Our “sneaking ligand” NF-kappaB inhibitor (named SLC1) inhibits NF-kappaB activation specifically in E-selectin expressing cells in vitro and in mouse models of arthritis indicating the importance of NF-kappaB in the activated endothelium. Clinical signs of arthritis were ameliorated by SLC1 treatment. We conclude that the SLFP architecture consisting of easily exchangeable domains represents an attractive approach to utilize other disease-relevant biological targets both on the cell surface and intracellularly. By relying on two independent disease specific targets, SLFPs may increase the therapeutic efficacy and reduce adverse effects.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease preferentially affecting the joints. Up to 1% of the world’s population is suffering from this chronic inflammatory disease that leads to dramatic joint deformity, loss of function and disability1.
Cellular interactions and a complex cytokine network (e.g., IL-1, IL-6, TNF-alpha, and IL-17) are involved in the development and progression of RA. The role of the individual cell types contributing to the pathogenesis of RA such as dendritic cells, B and T cells, monocytes/macrophages, neutrophils, fibroblasts and endothelial cells have already been well characterized. This understanding led to the development of cytokine inhibitors (e.g., etanercept, adalimumab, tocilizumab or anakinra) and cell-depleting agents (e.g., rituximab) for RA treatment2. Biologicals have significantly improved the treatment of RA, however adverse effects such as risk of infection limit their use, especially in patients with risk factors.
Recently, the Janus-kinase (JAK)-inhibiting small molecules tofacitinib and baricitinib got approved for the treatment of RA3.
Certain small molecule drugs can permeate the plasma membrane by passive diffusion and get access to cytosolic proteins to intervene in signaling processes for blocking pro-inflammatory gene expression4,5.
Also, cell-penetrating peptides (CPPs) that are consisting of a highly basic protein transduction domain (PTD)6,7 linked to an effector peptide represent an approach to modulate intracellular signaling. However, small molecule drugs and CCPs target every cell type and might influence homeostasis of the immune system8. The development of adverse effects with these compounds cannot be excluded.
Hence, detailed investigations to identify disease-relevant cell-types and signaling molecules for the development of innovative targeted therapies are required. In this mini-review, we illustrate a tool for cell-type specific modulation of intracellular signaling pathways that bases on multi-modular structured recombinant fusion proteins. We named these proteins SLFPs. The proof-of concept SLFP, named SLC1, elucidated the role of transcription factor nuclear factor kappa-light chain-enhancer of activated B cells (NF-kappaB) in the activated endothelium and ameliorated experimental arthritides9.
NF-kappaB: a key regulator of inflammation
NF-kappaB exhibits a broad role in regulation of target genes with various functions contributing to inflammation and immune activation, cell survival, and apoptosis10.
The NF-kappaB family in mammals comprises of five members termed p65 (RelA), RelB, c-Rel, p50 (NF-kappaB 1; and its precursor p105) and p52 (NF-kappaB 2; and its precursor p100) sharing a Rel-homology-domain (RHD) that allows DNA binding, dimerization and nuclear translocation. For gene activation, p65, RelB and c-Rel further contain the transcriptional activation domain (TAD). NF-kappaB/Rel proteins can exist as homodimers or heterodimers in any combination, whereas p65 and c-Rel preferentially interact with p50, and RelB with p5211.
Various stimuli activate the classical (canonical) or alternative (non-canonical) NF-kappaB pathway. IL-1 and TNF-alpha receptor signaling trigger the classical NF-kappaB pathway by phosphorylation of the IκB kinase complex (IKK complex) which is composed of the catalytic subunits IKK-1 and IKK-2 and the regulatory subunit IKK gamma (also designated as NEMO, NF-kappaB essential modulator)11. In resting cells, p50/p65 heterodimers are retained in the cytoplasm by the inhibitory binding of IkappaB proteins (Ikappa-alpha, Ikappa-beta and Ikappa-eta)12. Following IκB ubiquitination and proteasomal degradation, p50/p65 heterodimers translocate into the nucleus and bind to the promoter region of its target genes. Activation of NF-kappaB via the IkappaB kinase complex is critical for the expression of multiple pro-inflammatory genes including cytokines (IL-1, TNF-alpha, IL-6, IL-8), adhesion molecules like E-selectin, ICAM-1, VCAM-1, and P-selectin as well as chemokines like MCP-1 and IL-8 in primary endothelial cells13,14.
Dysregulated NF-kappaB activation contributes to the pathogenesis of RA as well as other pro-inflammatory conditions15,16. Furthermore, NF-kappaB activation is also involved in the resolution of inflammation and may exert positive or negative effects on inflammatory processes depending on the cell type and the disease phase17,18. Compelling evidence from experimental models of autoimmune diseases supports the concept of NF-kappaB blockade for therapeutic interventions19-21. We showed that NF-kappaB blockade in cytokine-activated endothelial cells prevents local recruitment of inflammatory cells to sites of inflammation9.
NF-kappaB inhibition
Targeting the NF-kappaB signaling pathway provides an outstanding tool for therapeutic intervention in immune-mediated inflammatory diseases and arthritis. To pursue NF-kappaB inhibition, different strategies can be applied.
Nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin and sodium salicylate, are common drugs in arthritis that prevents and dampen inflammation. Besides their functions to inhibit cyclooxygenases 1 and 2 (Cox-1 and Cox-2), enzymes that convert arachidonic acid into prostaglandins22,23, aspirin and sodium salicylate are competitive inhibitors of the ATP-binding site of IKK2. This event blocks IkappaB phosphorylation and inhibits NF-kappaB activation5. Furthermore, researchers already described in the 1990s that aspirin and sodium salicylate downregulate the expression of NF-kappaB target genes like VCAM-1 and ICAM-1 in endothelial cells and inhibit transmigration of leukocytes indicating a modulation of the NF-kappaB pathway by NSAIDs24. However, NSAIDs can cause severe side effects including renal failure, liver damage, and strong effects on the gastrointestinal tract and are not recommendable for patients with risk factors to NSAID-toxicity25.
NF-kappaB activation can also be controlled by oligonucleotide (ODN) NF-kappaB inhibitors. Antisense ODN’s were designed to NF-kappaB mRNA for controlling proinflammatory cytokine gene expression in lamina propria mononuclear cells (LPMCs) from patients with ulcerative colitis26. Successful amelioration of adjuvant arthritis was demonstrated using microencapsulated antisense NF-kappaB ODN27. Furthermore, NF-kappaB decoy ODN or small interfering RNA’s (siRNA) targeted against NF-kappaB components provide potential to control the transcriptional activity of NF-kappaB. In vitro experiments showed that NFkappaB decoy ODN inhibited cytokine and adhesion molecule expression in synovial cells derived from RA-patients28. However, clinical application of ODN and siRNAs is still hindered by lack of methods for organ or cell specific delivery, as well as their limited in vivo stabililty.
The pharmaceutical industry identified large numbers of small-molecule IKK2 inhibitors with anti-inflammatory capacities, also in collagen-induced arthritis29. However, so far none of the IKK2 inhibitors have been approved for clinical applications. Based on studies showing that inhibition of IKK2 results in liver cell apoptosis30 and embryonic lethality31, IKK2-dependent NF-kappaB activation plays an essential role in certain organs32.
Recently, approaches to inhibit IkappaB degradation and NF-kappaB nuclear transportation seemed to be more powerful, and may play a role in clinical applications. Bortezomib, a proteasome inhibitor, which inhibits the degradation of IkappaB, other regulatory proteins and misfolded proteins, depletes plasma cells and ameliorates clinical manifestations not only in NZB/W F1 mice, a mouse model for systemic lupus erythematosus (SLE), but also in refractory SLE33,34. Moreover, Bortezomib significantly improves severity and histological manifestations of collagen type II-immunized mice (collagen-induced arthritis, CIA)35.
Another option to block NF-kappaB activation is to inhibit the regulatory subunit NEMO of the IKK1/2/NEMO complex. May and collegues characterized a NEMO-binding peptide (NBP) that comprises the carboxyl-terminal segment of IKK2 to block NEMO-IKK2 association and inhibits cytokine-induced NF-kappaB activation and NF-kappaB gene expression36. Next, NBP was linked to the 16 amino acid long peptide penetratin, corresponding to the third helix of the Drosophila melanogaster homeodomain protein Antennapedia (Antp)37. Penetratin is known to pass through the plasma membrane38. The use of such cell-permeable peptide (CPPs) like Antp or a 10-amino acid long cationic stretch (amino acid sequence residues 47–57) of the 86 amino acid human immunodeficiency virus type 1 (HIV-1) TAT protein enables cellular uptake6,39 and represents an elegant approach for the transportation of an effector molecule into the cell interior40. However, the lack of cell specificity is a major drawback for the clinical application of CPPs. Also, protein size as well as hydrophilicity of amino acids limit the uptake into cells7,41. Antp-NBP interferes with the assembly of the cytosolic IKK complex and blocks the classical NF-kappaB pathway while leaving the basal NF-kappaB activity intact36,37.
Taken together, the findings mentioned above inspirated us to develop so called recombinant sneaking ligand fusion proteins (SLFP) to study the role of endothelial NF-kappaB activation in mouse models of arthritis.
Endothelium-specific NF-kappaB inhibition using the sneaking ligand approach
We developed an approach that allows cell-type specific modulation of signaling pathways and named this tool “sneaking ligand approach”9. The origin of these three-domain structured recombinant fusion proteins (sneaking ligand fusion proteins, SLFPs) lies in already described immunotoxins that target cells through specific single-chain variable fragments (scFv) or natural ligands that are linked to the translocation and effector domain of toxins like Pseudomonas Exotoxin A (ETA) or Diphteria toxin42,43,44,45. Hence, SLFPs consist of three domains: 1, the N’ terminal domain representing a binding domain, 2, the middle domain comprises the ETAII translocation domain that is linked to the 3, KDEL-containing C’ terminal effector peptide interacting with its cytoplasmic partner and modulating downstream signaling events (Figure 1a).
With regards to therapy, a crucial aspect for cell-selective modulation of signaling pathways is to target a cell-surface molecule or receptor that is preferentially expressed on cells that contribute to disease pathogenesis. In our study, we chose E-selectin as cell binding molecule, because this adhesion molecule is exclusively expressed under inflammatory conditions on endothelial cells. Therefore, the phage display derived high affinity peptide EBL (E-selectin binding ligand, AF10166 DITWDQLWDLMK) was introduced in our sneaking ligand prototype SLC146. The following ETAII domain47,48 facilitated the endosomal release of the c-terminal NBP into the cytoplasm to exert its function. Previously it has been described that upon receptor-mediated endocytosis of e.g. ETA and formation of early and late endosomes, ETA is guided via the RDEL /RDEL receptor interaction from the Golgi apparatus to the Endoplasmatic Reticulum (ER) in a retrograde manner. Cleavage of ETAII within the ER results in two fragments, thereby the RDEL-containing effector domain is released into the cytoplasm48. The mechanism of action of SLC1 is presented in Figure 1b. SLC1 is the first cell-type specific NF-kappaB inhibitor that exclusively blocks cytokine-induced NF-kappaB activation in the activated endothelium (Figure 1b)9.
In order to test the role of the individual domains for the function of SLC1, we generated mutants with a scrambled and deleted E-selectin binding domain, a scrambled NBP effector domain, a deleted ETAII domain and others9. The functional SLC1 and its non-functional mutants were expressed in E.coli9. The purified sneaking ligand proteins were used in in vitro and in vivo experiments to investigate the potential of SLC1 to inhibit NF-kappaB activation9: We showed by enzyme-linked immunosorbent assays and confocal microscopy that SLC1 binds specifically to E-selectin in in vitro cell culture system. First, a temperature-sensitive receptor-mediated endocytotic uptake was indicated by the intracellular distribution of SLC1 upon incubation at 37°C whereas experiments performed at 4°C exclusively resulted in cell surface staining9. The intracellular distribution of SLC1 in E-selectin expressing CHO cells (CHO-E cells) is depicted in figure 2. Second, transcriptional NF-kappaB activity was significantly reduced only in SLC1-treated E-selectin expressing cells. Third, we found a reduced NF-kappaB DNA-binding activity in an electromobility shift assay (EMSA), indicating that SLC1 inhibits the IKK complex activity in cytokine-activated E-selectin expressing cells and, thereby, prevents the translocation of NF-kappaB proteins into the nucleus. Forth, endothelial NF-kappaB blockade with SLC1 prevents cytokine-induced leukocyte trafficking in vitro and in vivo in a mouse model of acute thioglycollate-induced peritonitis. Fifth, we further showed by in vivo imaging that SLC1 binds to cytokine-activated vascular endothelium. Sixth, SLC1 decreased NF-kappaB activity and VCAM-1 in cytokine-activated mouse skin9. Finally, with regards to the therapeutic potential of SLC1, we investigated the impact of SLC1 in KxB/N serum transfer-induced arthritis (STIA) and antigen-induced arthritis (AIA)9. In STIA, serum of KxB/N mice which contains arthritogenic IgG antibodies against glucose-6-phosphate isomerase (G6PI) were transferred into naïve C57Bl/6J mice49. The formation of anti-G6PI-autoantibodies/G6PI immune complexes on the cartilage elicits Fc-receptor-mediated attraction and activation of neutrophils and mast cells and induces the release of pro-inflammatory cytokines like IL-1 and TNF-alpha. In AIA, a mono-arthritis is provoked by immunization with an emulsion of mBSA as antigen in complete Freund’s adjuvants followed by an intra-articular injection of the antigen in one knee joint cavity, whereas the other knee serves as control and receives PBS50. In contrast to STIA, AIA is a chronic, T-cell dependent arthritis model which is inducible in various mouse strains50. Moreover, a second intra-articular mBSA injection after 4 weeks results in a flare-up reaction of arthritis.
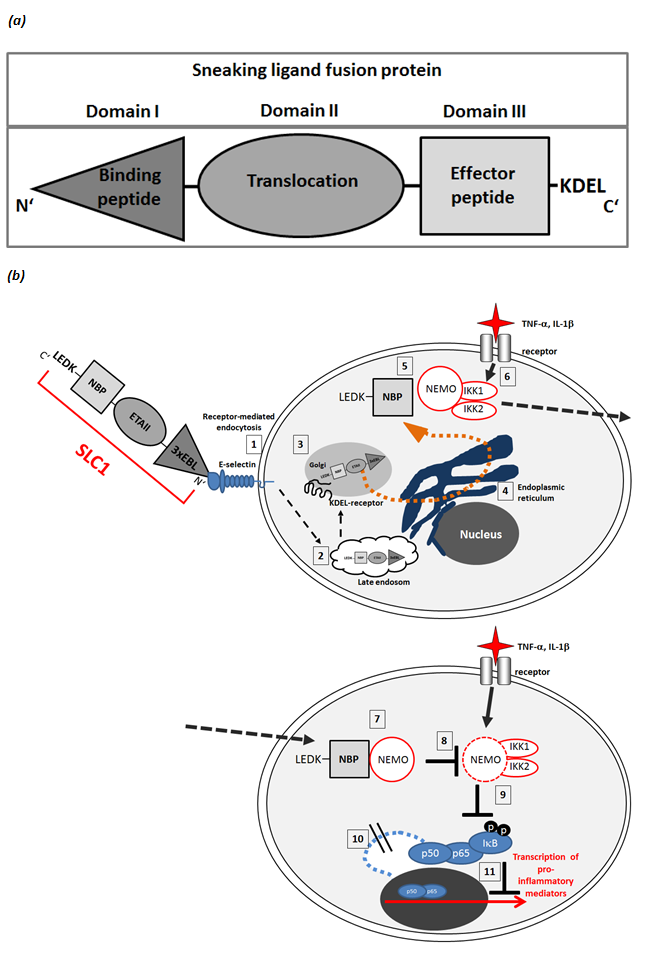
Figure 1: Structure of “sneaking ligand fusion proteins” and the “sneaking ligand” approach using the example of endothelium specific NF-kappaB inhibition9:
(a) SLFPs are composed of a three domains: I) the binding domain II) the translocation domain and III) the effector domain to interfere with intracellular signaling. (b) The endothelium specific NF-kappaB inhibitor SLC1 is composed of three repeats of the E-selectin–specific peptide (EBL, amino acid sequence: DITWDQLWDLMK)46, the translocation domain of Pseudomonas exotoxin A domain II47 and the NEMO-binding peptide (NBP) encompassing amino acids 644–756 from IKK237. A KDEL tag was added for improved intracellular trafficking. The domains are connected with S4G-linkers. 1, Binding of SLC1 to E-selectin induces receptor-mediated endocytosis. 2, Delivery of SLC1 into late endosomes 3, retrograde traffic of SLC1 from the Golgi to the ER via the KDEL receptor. 4 and 5, processing in the ER and release of NBP-effector domain into the cytosol.48 6, Cytokines activate the classical NF-kappaB pathway and induce the assembly of the IkappaB kinase complex consisting of IKK1/IKK2 and NEMO. 7, In the presence of SLC1, the NBP-effector domain interacts with NEMO and 8, the IKK complex (IKK1/IKK2/NEMO) cannot be formed. 9, Therefore IkappaB (inhibitor of kappaB) is not phosphorylated. 10, IkappaB is ubiquitinylated and degradated and, hence, p50/p65 dimers are not translocated to the nucleus. 11, in summary, SLC1 blocks the NF-kappaB-dependent transcription of several pro-inflammatory mediators.
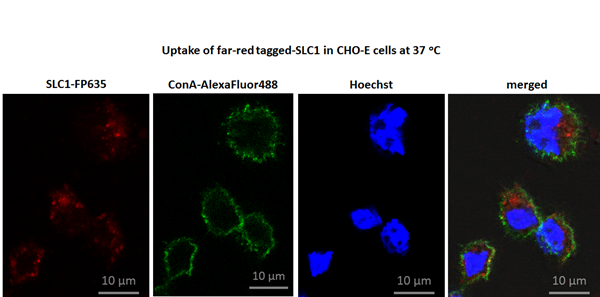
Figure 2: Intracellular distribution of SLC1 in E-selectin expressing cells (CHO-E cells).
FP635-tagged SLC1 (red colored) was incubated at 37oC with CHO-E cells for 1 hour. Plasma membrane was stained with Concanavalin A-AlexaFluor488 (ConA-AlexalFuor488; green colored) and nuclei were counterstained with HOECHST 33342 (blue colored). The merged image (right panel) shows intracellular distribution of SLC1. Imaging was performed using a Zeiss LSM 710 Axio Observer microscope9. CHO cells not expressing E selectin did not show any staining with SLC1 (not shown)9.
Here, endothelium-specific NF-kappaB inhibition ameliorated clinical manifestations in both mouse models of arthritis. SLC1 treatment reduced synovial inflammation and suppressed cartilage and bone breakdown in arthritic mice.
The importance of endothelial NF-kappaB activation is not restricted to inflammatory joint diseases such as arthritis but also known for other pathophysiological conditions including e.g., septic shock51, atherogenesis52 or tumor neoangiogenesis53. Hence, SLC1 may be also beneficial in these conditions. Importantly, selective NF-κB inhibition in cancer cells can sensitize them to chemotherapy54. Moreover, SLFPs might be useful in cancer therapy to manipulate deregulated cell proliferation and deregulated survival in a cell- or tissue-specific manner55.
A fundamental advantage of recombinant SLFPs is that both, the cell targeting and the intracellular effector domain, are exchangeable and therefore, different cell-types and/or signaling pathways may be easily addressed. However, SLFPs contain bacterial protein sequences, namely the ETAII domain and hence may be immunogenic, comparable to immunotoxins. Therefore, long-term treatments should be avoided. Immunogenicity can be decreased by using a shortened ETAII domain just containing the furin cleavage site48. Furthermore, instead of the artificial E-selectin binding peptide, natural ligands of mouse or human origin such as LFA-1 or cytokines can be used as non-immunogenic binding domains. We have recently successfully developed such constructs with low immunogenicity.
Recently, a slightly modified form of a SLFP was described as an “armed antibody” called DEKAVIL56. This recombinant engineered “armed antibody” is an immunocytokine composed of an antibody for tissue specific targeting fused to a cytokine but lacks the translocation domain used in SLFPs. DEKAVIL is an immunocytokine composed of the human antibody F8, specific to the extra-domain A of fibronectin linked to the human anti-inflammatory cytokine IL-10. F8 exhibits a strong affinity to cells from synovial biopsies56. In a phase IB clinical trial DEKAVIL showed first promising results on safety and reduction of disease activity and might be a novel therapeutic option targeting an immunomodulatory cytokine to the site of inflammation57.
Conclusions
The sneaking ligand approach represents a tool to study intracellular signaling in specific cell types and their role in certain diseases. SLC1, as an endothelium specific NF-kappaB inhibitor, revealed the crucial role of endothelial NF-kappaB in arthritis.
Sneaking ligands enabling a cell-type selective inhibition of defined signaling pathways may display an improved benefit-risk ratio compared to small molecule inhibitors affecting signaling pathways in all cells of the organism. Next generation SLFPs with reduced immunogenicity may be useful in the treatment of several human diseases including autoimmune diseases and cancer.
References
- Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016; 388: 2023-38.
- McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011; 365: 2205-19.
- Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Current opinion in chemical biology. 2016; 32: 29-33.
- Yang NJ, Hinner MJ. Getting across the cell membrane: an overview for small molecules, peptides, and proteins. Methods in molecular biology. 2015; 1266: 29-53.
- Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998; 396: 77-80.
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988; 55: 1189-93.
- Wadia JS, Dowdy SF. Modulation of cellular function by TAT mediated transduction of full length proteins. Current protein & peptide scienc., 2003; 4: 97-104.
- Senftleben U. Anti-inflammatory interventions of NF-kappaB signaling: potential applications and risks. Biochem Pharmacol. 2008; 75: 1567-79.
- Sehnert B, Burkhardt H, Wessels JT, et al. NF-kappaB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-kappaB in immune-mediated diseases. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 16556-61.
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008; 132: 344-62.
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes & development. 2004; 18: 2195-224.
- Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999; 18: 6867-74.
- Anand AR, Bradley R, Ganju RK. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-kappaB-dependent pathway. Mol Immunol. 2009; 46: 962-8.
- Collins T, Read MA, Neish AS, et al. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. Faseb J. 1995; 9: 899-909.
- Gregersen PK, Amos CI, Lee AT, et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet. 2009; 41: 820-3.
- Makarov SS. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001; 3: 200-6.
- Kanters E, Pasparakis M, Gijbels MJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003; 112: 1176-85.
- Lawrence T, Gilroy DW, Colville-Nash PR, et al. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001; 7: 1291-7.
- Herrmann O, Baumann B, de Lorenzi R, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005; 11: 1322-9.
- Jimi E, Aoki K, Saito H, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004; 10: 617-24.
- Voll RE, Mikulowska A, Kalden JR, et al. Amelioration of type II collagen induced arthritis in rats by treatment with sodium diethyldithiocarbamate. J Rheumatol. 1999; 26: 1352-8.
- Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature: New biology. 1971; 231: 232-5.
- Vane JR, Mitchell JA, Appleton I, et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91: 2046-50.
- Pierce JW, Read MA, Ding H, et al. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996; 156: 3961-9.
- Crofford LJ. Use of NSAIDs in treating patients with arthritis. Arthritis research & therapy. 2013; 15 Suppl 3: S2.
- Li Z, Zhang DK, Yi WQ, et al. NF-kappaB p65 antisense oligonucleotides may serve as a novel molecular approach for the treatment of patients with ulcerative colitis. Archives of medical research. 2008; 39: 729-34.
- Akhavein N, Oettinger CW, Gayakwad SG, et al. Treatment of adjuvant arthritis using microencapsulated antisense NF-kappaB oligonucleotides. Journal of microencapsulation. 2009; 26: 223-34.
- Tomita T, Takano H, Tomita N, et al. Transcription factor decoy for NFkappaB inhibits cytokine and adhesion molecule expressions in synovial cells derived from rheumatoid arthritis. Rheumatology. 2000; 39: 749-57.
- McIntyre KW, Shuster DJ, Gillooly KM, et al. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis and rheumatism. 2003; 48: 2652-9.
- Li Q, Van Antwerp D, Mercurio F, et al. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999; 284: 321-5.
- Tanaka M, Fuentes ME, Yamaguchi K, et al. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999; 10: 421-9.
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006; 25: 6758-80.
- Alexander T, Sarfert R, Klotsche J, et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015; 74: 1474-8.
- Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008; 14: 748-55.
- Lee SW, Kim JH, Park YB, et al. Bortezomib attenuates murine collagen-induced arthritis. Ann Rheum Dis. 2009; 68: 1761-7.
- May MJ, Marienfeld RB, Ghosh S. Characterization of the Ikappa B-kinase NEMO binding domain. The Journal of biological chemistry. 2002; 277: 45992-6000.
- May MJ, D'Acquisto F, Madge LA, et al. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000; 289: 1550-4.
- Derossi D, Joliot AH, Chassaing G, et al. The third helix of the Antennapedia homeodomain translocates through biological membranes. The Journal of biological chemistry. 1994; 269: 10444-50.
- Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988; 55: 1179-88.
- Strickland I, Ghosh S. Use of cell permeable NBD peptides for suppression of inflammation. Ann Rheum Dis. 2006; 65 Suppl 3: iii75-82.
- Lindsay MA. Peptide-mediated cell delivery: application in protein target validation. Current opinion in pharmacology. 2002; 2: 587-94.
- Bolhassani A, Jafarzade BS, Mardani G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides. 2017; 87: 50-63.
- Frankel AE, Fleming DR, Powell BL, et al. DAB389IL2 (ONTAK) fusion protein therapy of chronic lymphocytic leukaemia. Expert opinion on biological therapy. 2003; 3: 179-86.
- Allured VS, Collier RJ, Carroll SF, et al. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proceedings of the National Academy of Sciences of the United States of America. 1986; 83: 1320-4.
- Choe S, Bennett MJ, Fujii G, et al. The crystal structure of diphtheria toxin. Nature. 1992; 357: 216-22.
- Martens CL, Cwirla SE, Lee RY, et al. Peptides which bind to E-selectin and block neutrophil adhesion. The Journal of biological chemistry. 1995; 270: 21129-36.
- Wick MJ, Hamood AN, Iglewski BH. Analysis of the structure-function relationship of Pseudomonas aeruginosa exotoxin A. Mol Microbiol. 1990; 4: 527-35.
- Weldon JE, Pastan I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. The FEBS journal. 2011; 278: 4683-700.
- Ditzel HJ. The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med. 2004; 10: 40-5.
- Brackertz D, Mitchell GF, Mackay IR. Antigen-induced arthritis in mice. I. Induction of arthritis in various strains of mice. Arthritis and rheumatism. 1977; 20: 841-50.
- Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006; 290: L622-L45.
- Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis. J Clin Invest. 2001; 107: 255-64.
- Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008; 18: 19-26.
- Enzler T, Sano Y, Choo MK, et al. Cell-selective inhibition of NF-kappaB signaling improves therapeutic index in a melanoma chemotherapy model. Cancer discovery. 2011; 1: 496-507.
- Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001; 411: 342-8.
- Schwager K, Kaspar M, Bootz F, et al. Preclinical characterization of DEKAVIL (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthritis research & therapy. 2009; 11: R142.
- Galeazzi M, Bazzichi L, Sebastiani GD, et al. A phase IB clinical trial with Dekavil (F8-IL10), an immunoregulatory 'armed antibody' for the treatment of rheumatoid arthritis, used in combination wiIh methotrexate. The Israel Medical Association journal : IMAJ. 2014; 16: 666.