
The Impact of Nanoparticles on the Immune System: A Gray Zone of Nanomedicine
Priyanka Ray1, Noor Haideri2, Inamul Haque2, Omar Mohammed2, Saborni Chakraborty2, Snigdha Banerjee2,3#, Mohiuddin Quadir1#, Amanda E. Brinker 4,5, and Sushanta K. Banerjee2,3*
1Department of Coatings and Polymeric Materials, North Dakota State University, Fargo, ND
2Cancer Research Unit, VA Medical Center, Kansas City, MO
3Department of Pathology and Laboratory Medicine, University of Kansas Medical Center, Kansas City, KS
4Department of Cancer Biology, University of Kansas Medical Center, Kansas City, KS
5Institute for Advancing Medical Innovation, University of Kansas Cancer Center, Kansas City, KS
Abstract
Since the early days marking the first use of nanomedicine in the early 80s, there has been a meaningful change in the scientific field involving the Fabrication, characterization, and application of nanomaterials to treat many diseases, including cancers and genetic disorders. As unique and attractive properties of this novel class of materials unraveled, significant advances and discoveries were made over time. Addressing several challenges posed by conventional therapy, which were the only available treatment option for ailing patients, nanomedicine provided enhanced benefits, including reduced dosing, improved pharmacokinetics, and superior targeting efficiency. Several such formulations have successfully made their way to clinics and have shown promise in prolonging terminally ill patient populations' survival rates. However, the complex immune system and its various components, including various proteins and surface receptors, have made nanomaterials' journey from benchtop to the bedside a treacherous one. The innate and adaptive immune system interactions with nanomaterials are still under investigation and full of mysteries. This review highlights the various aspects of therapeutic nanocarriers and their current understanding of their immune systems' interactions.
Introduction
The field of research involving nanoparticles has invoked global interest due to their novelty and wide range of applications1,2. A nanoparticle (NP) may be defined as an assembly of atoms, molecules, or ions with a diameter theoretically between 1 and a few 100 nanometers with properties that differ from the atoms and the bulk. Figure 1 shows the nanoscale sizes of some species at different scales, starting from water which measures ~ 0.1 nm, to various biological species, including antibodies (~10 nm) and viruses and bacteria (100-1000 nm) and cancer cells which are larger than 1000 nm in size. Nanoparticles have attracted much attention in past decades due to their unique physicochemical and physiological properties, which differ significantly from their bulk form. Many of their physical properties like optical, magnetic, and catalytic properties, melting points, and surface reactivities are size-dependent, critical for all drug delivery applications from virus to human cells. This dependence is due to their high surface-to-volume ratio and their bridging state between atomic and bulk materials3. For any material to be used as a drug delivery agent, it is fundamental to study its immunological response in the body. Careful, rational design of nanomaterials and awareness of metabolic challenges are essential to avoid triggering undesirable immunological responses. Numerous clinical trials involving nanostructured materials have failed for the lack of a rational design4. Therefore, in this review, we set out to provide an overview of nanomaterials' interactions with one of those metabolic challenges, the immune system. We first provide a brief background of compositional and structural variability of nanomaterials found in the literature and their interactions with the immune system.

Figure 1: Size comparison of various objects on the nanometer scale (Created using BioRender). The ideal range of the size of the nanoparticle for drug delivery is 90 to 150 nm.
Classification of Nanoparticles
Nanomaterials are usually synthesized via various approaches and are usually classified as 'hard' or 'soft' based on their composition, size, and shape5. Tomalia, in his pioneering article, has provided a detailed description of this classification and a unifying systematic framework5. In many cases, hard nanomaterials are composed of noble metals like silver, gold, and platinum are an important class of nanomaterials owing to their unique optical and electrical properties. Therefore, these nanomaterials found applications in diverse fields, including sensors, medicinal biology, cancer therapy, optical filters, catalysis, and imaging. Metal nanoparticles have bactericidal properties, efficacy in treating cancer by photothermal and radiotherapy, and a single platform for therapeutic and diagnostic purposes, i.e., as theranostic agents6. The synthesis of these nanoparticles can be carried out by employing various chemical, photochemical, and radiolytic procedures. The synthesis method impacts the nanomaterials' properties, which can be tuned by choice of precursor and the technique employed. As metal nanoparticles tend to agglomerate during the growth and nucleation process, various ligands, stabilizers and supports control and limit their growth. Among these, polymers and micelles have been popular choices. Apart from the commercially available polymers, macrocycles, cyclodextrins, and calixarenes have emerged as a choice of ligands to stabilize metal nanoparticles7-9. However, due to certain metals' toxicity, their release into the biological media while imaging could be detrimental. Thereby, tampering with the toxicity of metal nanoparticles using organic stabilizers without compromising their salient properties is an active investigation area. Inorganic precursors, such as silicon, have been used to generate quantum dots (QDs) with enhanced photoluminescence for their use as contrast agents in various imaging applications. The development of QDs has been poised to change diagnostic and therapy landscapes due to their unique physico-optical properties. Still, these potent nanomaterials' inherent toxicity significantly limits their translational applications and requires further surface passivation, which sometimes leads to quenching of their luminescent properties.
Soft nanoparticles, which are the primary workhorse of nanotechnology-powered medicine, are derived from a host of precursors ranging from naturally occurring or synthetically designed lipids (liposomes)10, polymers (polymersomes, or micelles) 11, 12, dendrimers13, and other organic molecules14. These nanomaterials exhibit low toxicity, high biocompatibility, enhanced photoluminescence, and quantum yield, as well as substantial drug loading and programmed release capacities. These features render soft nanoparticles highly attractive for use in biomedical fields, especially for therapeutic applications (Table 1). Surface modification can enhance their aqueous solubility and allow for further conjugation with biomolecules that can be used as targeted therapeutic agents15.
Table 1: lists several nanoparticles used in drug delivery applications
|
Type of Nanoparticle |
Active Drug |
Disease Treatment |
Commercial Name |
References |
|
Liposome |
Doxorubicin Daunorubicin Amphotericin B Irinotecan |
Breast Cancer Leukemia Fungal infections Metastatic pancreatic cancer |
MyocetTM DaunoXome® AmBisome® MM-398 |
(16) (17) (18) (19)
|
|
PEGylated liposome |
Doxorubicin |
Ovarian and breast cancer |
Doxil®, Caelyx®, |
(20) |
|
Lipid nanoparticles |
Amphotericin B |
Fungal infections
|
Amphotec®, Abelcet® |
(21) |
|
Protein-based nanoparticles |
Paclitaxel |
Pancreatic, breast cancer, advanced non–small-cell lung cancer |
Abraxane® |
(22) |
|
Nanoemulsion |
Difluprednat Cyclosporine A |
Eye inflammation Dry eye syndrome, Keratitis
|
Durezol® Restasis®, Ikervis® |
(23) (24) |
|
Polymeric nanoparticles |
Sevelamer Leuprolide acetate
Recombinant antihemophilic factor |
Kidney disease Prostate cancer, endometriosis, uterine fibroids
Rheumatoid arthritis, spondylitis,
Crohn’s disease |
Renagel®, Renvela® Eligard®
|
(25) (26)
(27)
|
|
Nanpplatin(Nanocarrier) |
Polyamino acid, PEG, and cisplatin, Pembrolizumab |
Advanced solid tumors, lung, biliary, bladder, or pancreatic cancers |
KEYTRUDA® |
(29) |
|
CRLX101 |
Cyclodextrin-based nanoparticle-camptothecin conjugate |
Ovarian, renal cell, small cell lung, or rectal cancers 2016: NCT02187302 (Ph II): Completed |
(Cerulean) |
(30). |
Furthermore, the conjugation of fluorescent dyes and/or therapeutic drugs renders these nanomaterials attractive theranostic agents. Soft nanoparticles are frequently classified into two broad groups, including synthetic and biological. The first group comprises dendrimers, nanolatexes, and polymeric micelles, while the second is further subdivided into proteins, viruses, and DNA/RNA. The fundamental difference between hard and soft nanoparticles is their rigidity and degrees of freedom. A third category based on hybridized hard particle and the soft particle has also been identified, which can be a matrix of combinations between different types of hard and soft nanomaterials. Although nanoparticles have applications in a wide array spanning from electronics31 to biofouling32, our area of interest in this manuscript is in the field of nanoparticles used in cancer therapy and their interactions with the immune system. Therefore, in the following sections, we provide a detailed overview of nanoparticles heavily investigated in cancer therapy, such as metal nanoclusters, liposomes, and polymeric nanoparticles, as representative examples of hard and soft nanomaterials usable in medicine and diagnostics (Figure 2).
Metal nanoclusters
Noble metal nanoparticles have been used to heal diseases like measles and ulcers since historic times. Although the preparation of metallic colloids dates back to the middle ages, the history of metal nanoparticles began with Faraday’s study of gold colloids33. In contrast, most metallic nanoparticles are used in electronics and semiconductors, and noble metal NPs have been widely used in the medicinal field. The bactericidal properties of silver, the anticancer properties of gold compounds, and a host of platinum-containing chemotherapy drugs have made them much sought after in the field of nanomedicine.
Like most other nanosystems, synthetic routes of metal NPs broadly involve either top-down or bottom-up methods. The top-down methods are used to prepare NPs via the subdivision of bulk metals, including condensation/evaporation, pyrolytic methods, laser arc discharge, lithography, and electronic radiation. Simultaneously, the bottom-up processes generate metal NPs from the zero-valent metal using molecular or ionic precursors under the conditions that prevent their precipitation. Techniques like molecular self-assembly, atomic layer deposition, reduction using chemical or irradiation are employed for the bottom-up method.
Given that dimensions of metal clusters or metallic nanoparticles are comparable to biomolecules [such as a virus (∼ 20 nm), proteins (1–20 nm), and DNA (∼2 nm)], they are a popular choice for use to interact or intervene with different biological systems. Such engagement can be used for therapy34 and the detection of diseases. For example, the nanoscale dimension of nanoparticles permits their interactions with biomolecules on cell surfaces and within the cells in a way that can be decoded and designated to interact with critical biochemical and cellular processes. When bound with the drugs, they can penetrate the tissues and cells in the body and give a target-based drug therapy, which is highly selective and specific35. Nanoparticles have also been employed to mimic biological systems to create better drug carriers and sensors. Due to the high surface to volume ratio of nanoparticles, functional groups' multivalency can be achieved and used to modulate these nanomaterials' interactions with cells36. A primary concern while using metal nanoclusters is the toxicity of the metals themselves and their cellular or organ-level accumulation. For metallic nanoparticles, surface reactivity, size, and shape have been established to drive the compromise between their therapeutic and toxic impact37.
Liposomes
Liposomes are one of the oldest members of the soft nanomaterials family (Figure 2). These species are generated using phospholipids comprising a hydrophilic head and hydrophobic tail. When using aqueous solvents, they form vesicle-like structures capable of entrapping therapeutics in their core. Various hydrophobic and hydrophilic interactions between the lipids themselves and lipid-water lead to bilayered structures. Since phospholipids form the backbone of cell membranes and plasma lipoproteins are natural lipid NPs, liposomes are biocompatible and usually present low to no toxicity.
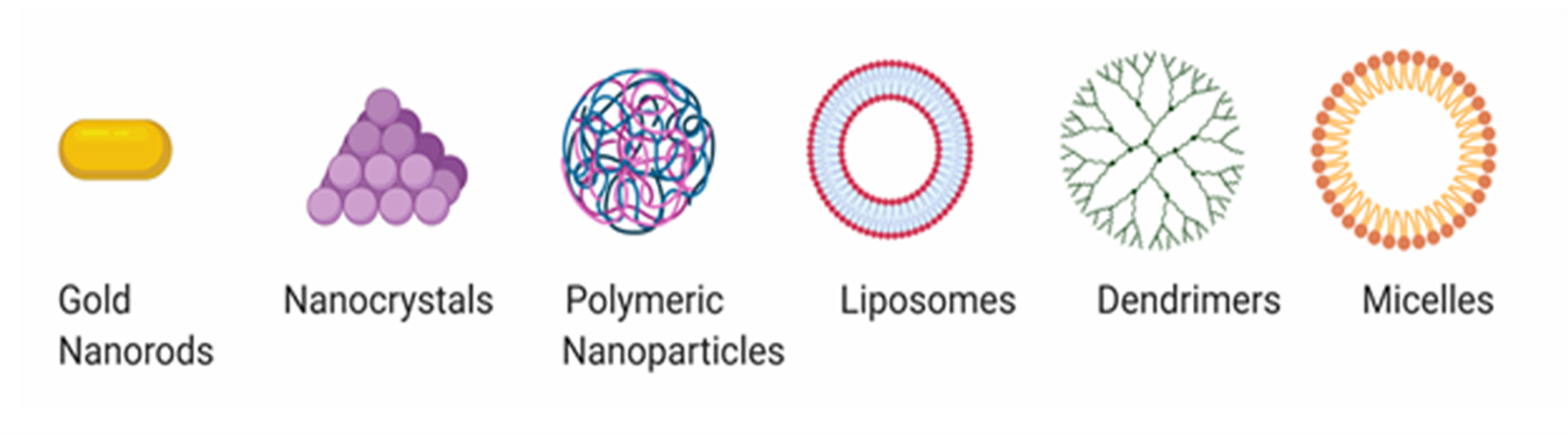
Figure 2: Representation of some widely used classes of nanomaterials in cancer therapy. (Left to right). Gold nanorods, which are cylindrical particles derived from gold metal salts, metal nanoclusters, usually a clump of metal nanoparticles stabilized by various ligands, polymeric nanoparticles, derived from a host of polymers via synthetic methods, liposomes which are bilayer structures from lipids, dendrimers, branched structures with multiple arms and micelles which have a hollow cavity with a distinct head and tail dependent on the hydrophilicity of the components. (Created using BioRender)
Based on their size and layer number, liposomes are classified as multilamellar vesicles, large unilamellar, and small unilamellar vesicles. Based on composition, they are categorized as conventional, pH-sensitive, cationic, long-circulating, and Immuno-liposomes38. The delivery-based classification includes-conventional, sterically-stabilized, ligand-targeted liposomes, and a combination thereof.
The use of liposomes has vastly improved the therapeutic index of several drugs, including Doxorubicin and Amphotericin39. This is achieved by improving the pharmacokinetics and biodistribution. However, they are often rapidly eliminated from the bloodstream due to the opsonization of plasma components and macrophages' internalization in the reticuloendothelial system (RES)40. PEGylation polymers are a popular approach to address this issue. PEG chains increase the concentration of hydrated groups on the surface, which sterically inhibit both electrostatic and hydrophobic reactions with plasma proteins, thus reducing the internalization of the formulation by the RES41. Since the size of liposomes varies largely, this factor also impacts their interactions with the immune system. Larger liposomes are prone to more rapid clearance than smaller ones.
Along with size, the surface charge is also determinant in governing liposome fate. More prominent and charged liposomes were found to be eliminated rapidly by the liver and spleen. Using cholesterol in the membrane has prolonged the circulation time of liposomes42. Liposomes that have not been cleared by RES and opsonization are subjected to the enhanced permeation retention (EPR) effect, which governs the vascular supply's increased permeability to cancer tissues. Liposomes can be tailored to accumulate by passive targeting using various triggers like pH, hypoxia, and temperature43.
Polymeric nanoparticles
Many biodegradable polymers have been used as drug delivery agents and have increased therapeutic efficiency with minimal off-target toxicities44. Several specialty polymers like poly (lactic acid) (PLA), and poly (lactic-co-glycolic acid) (PLGA) have got considerable attention due to their biocompatibility with and controlled release properties of chemotherapeutic agents45. Other polymers derived from naturally occurring sources like sodium alginate and chitosan46 have also been employed as drug delivery agents. Lipid or protein-based soft nanoparticles comprises another important class of soft nanomaterials. Therapeutic liposomes are composed of lipids and are best represented by DOXIL®, an FDA-authorized product for soft tissue cancer. Another clinically relevant polymer, Abraxane®, an albumin nanoparticle of a potent chemotherapeutic agent (paclitaxel), is a clinically used nanomedical formulation for cancer therapeutics.
Apart from the commercially available polymers mentioned above, a host of synthetic biodegradable polymers has been synthesized and can efficiently deliver therapeutics at the desired site. For such transport to the disease site, two types of targeting mechanisms are generally followed: (a) Passive or size-dependent transport and (b) Active or ligand-mediated transport. Microenvironment-sensitive chemical units have also been immobilized onto both soft and hard nanoparticles to sense biochemical triggers like hypoxia, pH, enzyme concentration, and temperature. These stimuli-sensitive polymers form self-assembled structures at the physiologically prevailing conditions but can sense the specific trigger distinctive to the cancer microenvironment. Such sensory input is translated to the collapse of self-organized structures leading to drug release. Significant advancements have been achieved in the area of stimuli-responsive polymeric nanoparticles for cancer therapy47, 48. Inspired by these works, we have reported the synthesis and use of several pH-responsive block copolymers comprising hydrophilic PEG and hydrophobic polycarbonates for delivery of newly discovered molecular inhibitors, such as hedgehog inhibitors49 (GDC 0449), ERK inhibitor50 (SCH 772984) or conventional chemotherapeutic agents such as Gemcitabine. Physical encapsulation or chemical conjugation of these molecular species within stimuli-sensing nanoparticles improved their therapeutic profile by protecting the drugs against rapid metabolic clearance and suppressing their off-target toxicity51. Although physical encapsulation of drug molecules inside polymeric nanocapsules is a popular and facile strategy for designing controlled-release drug delivery systems, chemical conjugation to polymeric scaffolds has also been widely employed by researchers for designing polymer-drug conjugates. Conjugation of drugs is achieved via chemical coupling of drugs to the polymer backbone via simple amide or ester bond linkage, which, when subject to certain conditions prevailing in the cancer tissues, leads to their cleavage subsequent release of the drug52, 53. This approach protects the drug against enzymatic degradation, prolongs its circulation time, and ensures efficient payload delivery to the targeted tumor tissue54-59.
Soft nanoparticles have been equipped with targeting ligands, immobilization of which on nanoparticle surface is also a popular strategy to design "ligand targeted therapeutics." Several ligands, including peptides, polysaccharides, proteins, aptamers, and small molecules, have been used for targeting the tumor tissue. The rationale behind employing targeting ligands is to harness their binding affinity to specific receptors on the cell surface, thereby increasing the therapeutic efficiency and accumulation of the NPs on a specific population of cells. As this is mostly a surface phenomenon, it is frequently more advantageous to employ multiple ligands instead of a single one to increase cellular uptake via a multivalent effect60 (presence of the multiple numbers of functional ligands). Multivalency is concerned; dendrimers and hyperbranched polymers have been developed and are included within the family of soft nanomaterials.
Dendrimers are a class of materials that are branched at the edges and possess a unique 3-dimensional structure. These are usually classified as macromolecules or branched polymers and have been useful as novel drug delivery agents. The structure of a dendrimer is usually in the form of an inner core, with each progressive shell or branching out referred to as a 'generation.' With the increase in the shells' branching (ie: increase in the generation numbers), the molecular mass also increases. Multivalent functional groups, including -COOH, -OH, and -NH2 are often used for interactions with other chemical agents or biological molecules. The core-shell structure of dendrimers that gives them macromolecular properties has often been used for 'host-guest' type interactions for sensing of various ions or biomolecules and entrapment of drugs and dyes. Some commercially available dendrimers of polyamidoamine (PAMAM) (Starburst®), Poly(propylene imine (Atramol®), and Polylysine (Vivagel®) are already available commercially for medicinal applications. Synthesis of the perfect dendrimer is often cumbersome, and therefore more easily realizable dendritic scaffolds have been developed. Termed as 'hyperbranched polymers,' these nanoscale unimolecular species demonstrate spherical presentations of dendrimers; however, they do not require a stepwise synthesis to realize a multivalent and branched topology.
As with polymeric nanoparticles, various active and passive targeting strategies have been applied to dendritic systems to render them highly effective in delivering therapeutics to the tumor core with minimal off-target toxicity. Particularly with non-polar chemotherapeutic agents, dendrimers and hyperbranched polymers provide a useful scaffold to aid in solubility and formulation stability of chemotherapy without compromising their therapeutic efficiency61-63. Conjugation of specific antigens for specific delivery using monoclonal antibodies has also been explored64 as applications of dendrimers and hyperbranched polymers for vaccine research.
Nanoparticle Mediated Immunotoxicity
Since the advent of nanomedicine, there has been a surge in the use of various nanomaterials described in the previous sections for a whole range of therapeutic applications. Although several features of nanoparticles in terms of high payload, low dosage, and targeting capacity make them attractive candidates for use in cancer treatment, an essential aspect of clinical consideration remains the Immunotoxicity of these NPs.
When used for in vivo application, especially for systemic delivery of therapeutics, NPs, and biological agents' interface play a fundamental role in nanomaterials' fate in effective delivery, clearance, and accumulation.
The immune system's primary function is to protect the body against foreign particles considered threats65. The broad classification of the immune system is innate and adaptive. Innate immunity generates a non-specific inflammatory response when in contact with foreign bioactive agents like bacterial and viral strains. This system's mechanism of action is through various receptors functioning as pathogen recognition agents followed by the acquired immunity system's activation through antigen-presenting cells (APCs). In contrast, the adaptive immune system is responsible for generating antibodies in response to the antigens and follows a complicated pathway. Immune systems recognize nanoparticles via their surface properties and compositional features. Therefore, these features can act as handles to mediate interactions of nanoparticles with the immune system.
The first contact of NPs with biofluids exposes them to various proteins to form a crown effect commonly referred to as protein corona particle66. Although other biomolecules like DNA, RNA, and ribose sugars also interact with NPs, they have been underrepresented in literature and ensuing scientific investigations. Interactions of the NPs with the plasma proteins and other biomolecules alter their fate as various biochemical changes alter the bioactivity of NPs.
Phagocytes, which comprise an essential part of the immune system, are the first to interact with any foreign bodies, including nanoparticles. The two types of interactions this brings about are immunosuppression or immunostimulation. While immunosuppression renders the host organism susceptible to various infections due to the immune system's incompetency to combat any invasion, immunostimulation is responsible for a host of disorders, including inflammation.
Undesirable interactions between the immune system and nanoparticles have often been reported due to immunostimulation or immunosuppression (Figure 3), which might cause inflammatory or autoimmune disorders, thereby increasing the chances of the receptor's body incurring an infection. Immune recognition can be bypassed via several methods, of which using a polymeric substance to create a hydrophilic environment is amongst the most popular. However, this cannot negate antibody production. On the other hand, to elicit the desired immune-response, antigen-presenting cells are directly stimulated, or antigen is delivered to the desired cellular compartment via targeting methods.
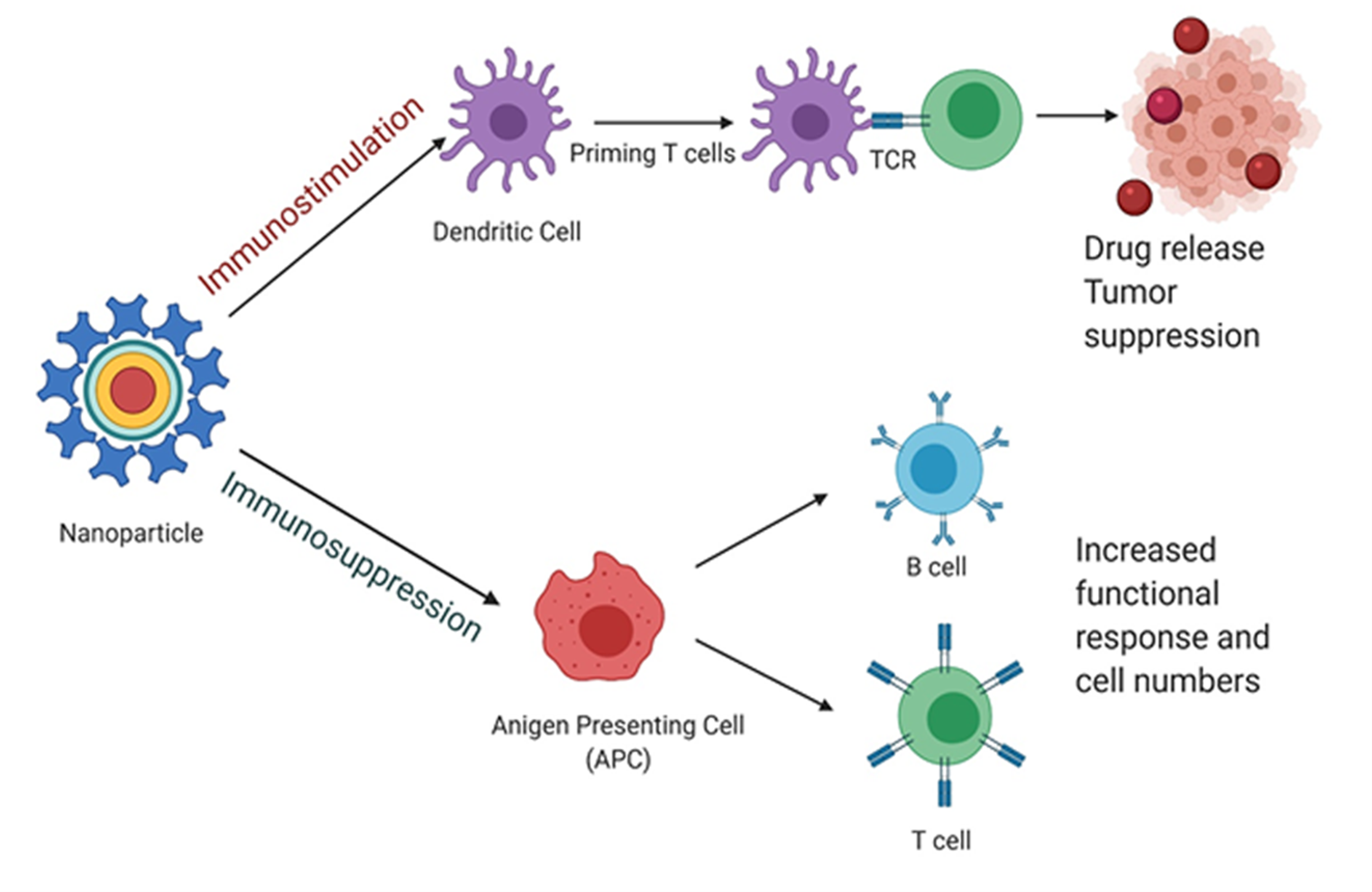
Figure 3: Schematic representation of the two significant modes of interactions between nanoparticles and the immune system. TCR (T-cell receptor) These modes are (i) immunostimulation of dendritic followed by priming of T-cells, eventually leading to drug release into the tumor cells and (ii) immunosuppression of APC, which increase in T and B-cell numbers and functionality. (Created using BioRender)
Immunosuppression via nanoparticles has been achieved in several ways, and one such method was via the inhalation of carbon nanotubes (CNTs) to suppress B cell function. The cytokine transforming growth factor-β (TGF-β) produced by alveolar macrophages is a critical element of the observed immunosuppression mechanism.
On the other hand, for nanomedicine administered via subcutaneous or intradermal routes, activation of the complement by the nanoparticles can enhance the treatment's efficacy. Several systems have been reported to improve antigenicity of conjugated weak antigens and thus serve as adjuvants, while engineered nanosystems have also shown inherent antigenic properties. Nanoparticles activate the complement system via different pathways, and by changing their physicochemical properties, this activation can be regulated. Mast cells can contribute to inflammation and some nanoparticles' toxic effect and have an essential role in the biological events following nanoparticle exposure67.
A host of factors, including size, crystallinity, aggregation properties, composition, shape, and surface charge, influence the interactions between the immune system and nanoparticles (Figure 4).
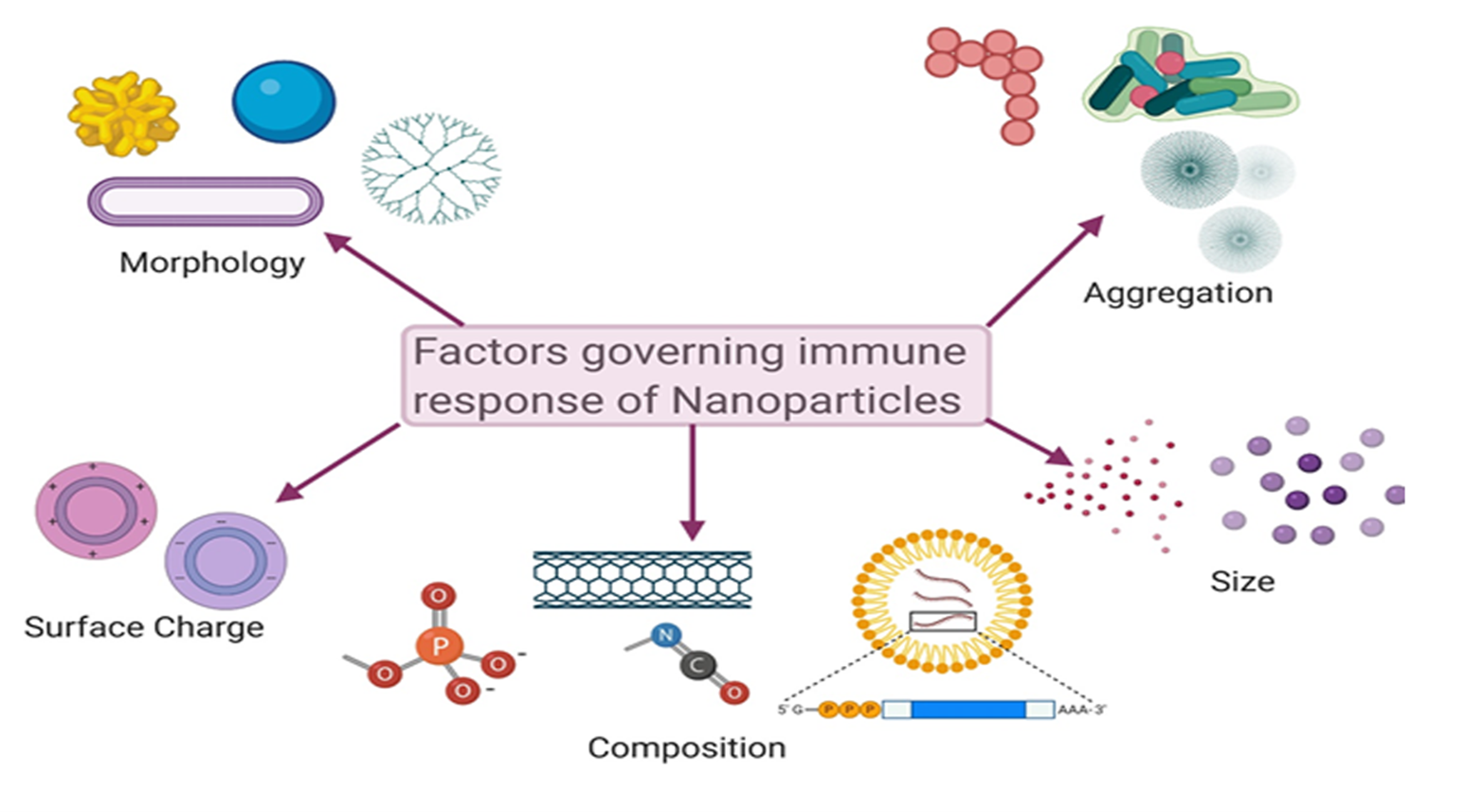
Figure 4: Schematic representation of several factors governing the immune response elicited by nanoparticles. These factors are morphology, size, shape and surface charge of nanoparticles and their chemical composition and aggregation properties. (Created using BioRender)
Impact of Nanoparticles in vitro and in vivo on the Immune Response
Uptake by the innate immune system transfers NPs within a recognizable size range to these areas, where the lymphatic system directs them. In vivo, murine models show adverse effects in these areas and suppress natural killer (NK) cells68. These models have also shown a variety of interactions with T and B cells in these areas. Models involving negatively charged SiO2 showed suppression of these cells, inhibiting NK cell activity, suppressing proinflammatory cytokine production, resulting in a lack of inflammatory responses68. Silver nanoparticles (AgNPs) are bound to various tissues, resulting in reactive oxygen species (ROS) induction, inflammation, and tissue damage. The toxic effect of AgNPs on the proliferation and cytokine expression by human lymphocyte cells and peripheral blood mononuclear cells (PBMCs) has also been investigated69. The immunosuppression induced by these AgNPs caused broad organ damage in these areas and harmed organisms. The accumulation of NPs can also harm organs like the liver, spleen, and lymph nodes.
Despite the disconnect between in vitro and in vivo models, RAW 264.7 cells (murine macrophage-like cells) are the most commonly used cellular model for immunotoxic assays. These cells show pronounced dose-dependent cytotoxicity. NPs are best dosed out by a surface area factor, not mass, since the surface area is the biologically most effective dose metric for acute nanoparticle toxicity. When proportionally applied to cell cultures, silicon NPs caused more significant cytotoxicity than controls to monocytes and macrophages70. This toxicity was evidenced to be the product of ROS release and oxidative stress. These NPs also contributed to most lymphocytes' death through apoptosis and necrosis, which causes direct cellular damage.
Despite the apparent disconnect between in vitro and in vivo models, results have been consistent. The leading cause of cytotoxic factors created by NPs is TLR activation, ROS production, and the triggering of direct proapoptotic factors. Broad immunosuppression and accumulation can also lead to organ and cell damage. These issues can potentially be offset by immunomodulatory practices that would breed tolerance within the immune system.
Correlation between in vitro and in vivo Toxicity
There are various reasons that the correlation between in vitro and in vivo toxicity can be inaccurate. This is because the distribution of nanoparticles across a surface area may differ. Different cell types do not uptake all nanoparticles the same way. The immune system may also remove some of the nanoparticles before reaching the target area, so cells may not receive the same nanoparticle dose as expected. For example, in a study comparing 2D and static in vitro conditions to a 3D culture, the heterogeneous distribution patterns in the 3D culture were similar to those in vivo.
In contrast, the 2D models showed an equal distribution. This is because 3D cultures are similar to the form of tissues in the body. They showed less toxicity in comparison to the monolayer cultures.
Another way to more accurately display in vivo conditions is through the use of dynamic flow models. Nanoparticle interactions with endothelial cells can be more accurately recapitulated because they are maintained under flow conditions (instead of static cultures), leading to Immunotoxicity's suppressed extent. Thus, when a nanoparticle is under flow conditions, it does not come into as much contact with cells. Therefore, the intracellular uptake was reduced, thereby reducing the toxicity.
Size and Structural Importance of Nanoparticles in Immunological Considerations
A host of factors determine the fate of NPs with regards to their capability of either deceiving or being detected by the immune system. As discussed earlier, size, shape, and surface properties are among prime components that control the immune response of the NPs.
The innate immune system recognizes foreign bodies based on their size, amongst others. Thus, NPs must be engineered to be of a specific size range to avoid being rejected as 'foreign' bodies. As the size increases, it causes a decrease in the surface-to-volume ratio of NPs, thereby affecting their surface interaction with various immunoreagents. The main processes through which NPs are taken up by cells include pinocytosis, macropinocytosis, phagocytosis, clathrin, or caveolar-mediated endocytosis71, as well as the removal of lipids by the lipoprotein receptor, SCARB172, 73. Amongst these, the first two-act by non-specific responses. Large-sized NPs usually interact with APCs abundant on the tissues, whereas NPs with sizes < 200 nm usually circulate through the venous and lymphatic drainage and increase antigen presentation74. NPs with sizes ~ 50 nm were found to increase the expression of certain cell markers, including CD40 and CD80, inflammatory cytokines like IL-6 and TNF-α, which are hallmarks of a primary response by the immune system75. Another major hurdle in the employment of NPs is their retention in the liver, which is mostly governed by their size, as the accumulation increases for the larger size. On the other hand, smaller NPs are cleared rapidly by the lymphatic system to be taken up by resident dendritic cells76.
Apart from the size, the shape of NPs is also a governing factor for an appropriate immune response. Reports have indicated that rod-shaped NPs with a larger surface area than spherical NPs were much more likely to be taken up by macrophages. This was attributed mainly to the generated vesicles, which allow for easier engulfment via micropinocytosis. For rod-shaped structures, the aspect ratio was reported to govern cytokine secretion77; the larger the aspect ratio, the more enhanced the production of certain inflammatory markers like IL-6 and IFN- γ. Researchers have also conducted studies on complement activation by maintaining the same surface area but changing the nanoparticles' shape. Results indicated that although all types of NPs were capable of inducing activation of C3a and C5a complements as soon as they came in contact with blood, elongated rods had a more pronounced effect than spheres78.
Since the NPs surface is the first point of contact with the immune system, a detailed investigation of their surface properties would help advance the field of nanoparticle-based Immunotoxicity to a great extent. Hydrophobic functional groups in pathogenâassociated molecular pattern molecules (PAMPs) that participate in danger signaling pathways can vastly modify the nanocarriers' fate. NPs with a higher hydrophobic surface was rapidly internalized and induced higher expression of CD8679 and decreased binding of immunoglobulins and apolipoproteins on the NP surface. The cationic surface charge is also responsible for inducing binding with various plasma proteins and forming the famous 'protein corona,' which is then taken up by macrophages. For zwitterionic NPs, the uptake is lesser than cationic NPs as there is a lower accumulation of serum protein on their surface80.
As discussed earlier, the "protein corona" or PC is marked as a recognized foreign entity by specific receptors, including TLR and complement receptors81. A process by which complement proteins adsorb onto the NP surface is known as 'opsonization,' which allows for the clearance of the NPs by the immune system. This is usually addressed using polymers like PEG or PEGylation of the NPs to create a hydrophilic layer that would inhibit protein adsorption by blocking binding sites. Blocking binding decreases protein adsorption and protects the NP and its therapeutic payload from the immune system's rapid clearance, thus enhancing their circulation half-life82.
Along with surface functionalization, the surface charge of nanocarriers influences their interactions with the immune system significantly. NPs with a positive surface charge increase the electrostatic attraction between NPs and negatively charged cell membranes, causing enhanced surface endocytosis83.
Surface modification of nanocarriers targeting ligands modifies the phagocytosis pathway, a primary part of the innate immune system. Phagocytosis involves competing for pro and anti-phagocytosis mechanisms, governed by the binding of signal regulatory protein SIRPα present on APCs with CD47 transmembrane proteins84. Using CD47 proteins to decorate the NP surface would thereby affect this signaling pathway. Similarly, liposomal formulations mimicking lipoproteins were reported to inhibit TLR-4 dependent inflammatory response by interacting with LPS85. Metal NPs, known for their toxicity due to the accumulation of metals in vital organs like the liver, have been reported to influence the regulation of inflammatory cytokines and upregulate levels of IL-6, TNF-α, and others in certain cell lines86.
Liposomes, a class of popular drug delivery agents, have often been reported to trigger an immune response originating in the innate immune response, thus causing an acute hypersensitivity syndrome known as complement activation-related pseudoallergy (CARPA). Clinically approved doxorubicin formulations like DOXIL have also been reported to cause CARPA87. This is a severe hypersensitivity reaction that occurs immediately post-injection and manifests itself with symptoms including swelling, chills, and anaphylaxis, among others. Thus, the use of Doxorubicin formulations is strictly monitored for patients with cardiac conditions and has also been reported to cause acute cardiotoxicity. This pseudoallergy is attributed to the activation of the complement system and anaphylatoxins generation87. The binding of anaphylatoxins to macrophages starts a cascade of processes, including histamine release, prostaglandins, and platelet-activating factor (PAF). Complement activation largely depends on the size, shape, surface charge, packing density, morphology, and functionalization of the liposomes. Small, non-charged unilamellar structures have been reported to cause a minimal reaction. A host of approaches are being pursued to reduce the immunotoxic response generated by liposomes as drug delivery systems.
Selection Criteria of Immunotolerance of Nanoparticles in Preclinical Models
Certain drugs often elicit unwanted immune responses that will lead to the production of anti-drug antibodies (ADAs). ADAs have an increased risk of immune-related adverse events, including infusion reactions, accelerated drug clearance, allergic reactions, life-threatening anaphylaxis, autoimmunity, and loss of response to the drug88. Inhibition of ADAs production in an antigen-selective manner is a high unmet need to reduce clinical failure of novel biologics in development and improve existing drugs' safety and efficacy. Researchers from Selecta Biosciences in Massachusetts have been working to prevent ADAs production with a nanoparticle-based platform called ImmTOR that can be shown to induce immunotolerance with ImmTOR nanoparticles encapsulating rapamycin, an immunomodulator89. A recent study, a highly immunogenic antigen, keyhole limpet hemocyanin (KLH) alone, synthetic vaccine particles containing rapamycin (SCV-Rapamycin, now known as ImmTOR) alone and concomitantly with SCV-Rapamycin were administered intravenously to mice then challenged with KLH alone. Mice were then checked for the production of anti-KLH antibodies. Their results suggest that KLH + SCV-Rapamycin was efficacious in inhibiting anti-KLH antibody responses. PLGA-NPs are the most frequently assessed Immature dendritic cells (DCs) treated in vitro with PLGA-NPs encapsulating dexamethasone (Dex), a potent immunosuppressive and anti-inflammatory drug, and a model antigen ovalbumin (OVA) did not mature into immunogenic DCs but instead were transformed to tolerogenic DCs90.
Furthermore, intragastric feeding and intravenous injection of these nanoparticles loaded with Dex and OVA induce OVA-specific immune tolerance in mice. Negatively charged poly(lactide-co-glycolide) nanoparticles encapsulating gliadin protein (TIMP-GLIA) were developed and tested for the immunomodulatory treatment of celiac disease91. TIMP-GLIA significantly reduced markers of gliadin-specific T cell activation, inflammatory cytokine secretion, and tissue damage when injected into mice's blood in celiac disease models. Gliadin nanoparticle treatment also induced FOXP3 and gene expression profiles associated with immune tolerance. These findings support the reprogramming immune system's concept to instruct T lymphocytes to tolerate gluten again in celiac patients. Similar nanoparticles may also be developed for the treatment of other autoimmune diseases.
The selection of immune-tolerant NPs is based on various design and end application criteria. It depends on many factors such as (i) the size of the desired nanoparticles to allow for efficient transport to lymphoid organs and targeting of DCs, (ii) the use of approved biocompatible and biodegradable polymers licensed for multiple uses in clinical settings, (iii) use of a small-molecule immunomodulator that has been validated in humans and is capable of inducing immune tolerance programming in DCs and antigen-specific Tregs, and (iv) co-delivery of antigens with the immunomodulator and delivery of instructions to antigen-presenting cells.
Approaches for Translation of Mouse-data to Humans in the Context of Immunology
Limitations of using mouse-model to generate immunological data associated with nanoparticle therapy
Mouse models have been the cornerstone of evaluating immunotoxic effects of nanoparticles designed to achieve therapeutic benefits against different disease conditions. In the context of developing cancer therapy, this is even more critical, as the information from rodent experiments forms the basis of go/no-go decisions for non-human primates (NHP) or clinical models. Although in vivo experiments with mouse models generate reliable data in evaluating the efficacy of anticancer therapeutics' safety, genetic, molecular, and physiological interactions, limitations in these models result in less consistent predictions for human clinical trials. Mice are limited as models for most human diseases, including cancer. This is because the pool of critical molecular, immunological, and cellular features of human cancers is absent in the mouse model. Gawrylewski et al. reported that among 4000+ genes in humans and mice, the binding site for transcription factors differed between the species in 41 to 89% of cases92. Although the mouse model can recapitulate a specific or a set of cellular processes of human disease, it cannot capture the global spectrum of physiological changes that occurs during that pathological setting.
Origin of failure
Of many reasons why mouse models might present a disconnect between preclinical and clinical data, immunological factors have been considered the most potent ones93. Reports suggest that specific pathogen-free (SPF) husbandry, a common practice adopted across laboratories conducting mouse experiments, has a broad and unexpected effect on the mouse immune system94. Generally, in most cases, SPF mice have an immature immune system compared with wild strains of rodents or mice95. Therefore, the immune system of SPF mice does not adequately reflect that of adult humans. For example, human and mouse physiology is influenced by the microbiome associated with various diseases, including cancer96. Influence of bystander infections97, co-infections, and the overall change in metagenome (i.e., the sum of host and microbial genes) drives the host's phenotypes for a given disease condition. It thereby leads to the significant differences in immune-status of individual species98. Bureau et al. showed that the immune response of laboratory mice bred and developed in an SPF facility has a significant difference from that of wild or mice purchased from a pet store95. While wild or commercially procured mice were enriched in differentiated memory T cells in lymphoid and nonlymphoid tissues, laboratory mice severely lack differentiated memory T cells. When gene expression was compared from peripheral blood mononuclear cells (PBMCs) from feral and laboratory mice with human cord PBMCs and adult PBMCs, gene signature was similar for wild mice and humans. These data indicate that environmental exposure shapes and alters mouse immune-responses, and therefore a considerable difference in immune-status exists between SPF-mice and adult humans. A challenging feature of humanized mouse models created for human cancer study is that the inoculated human tumors show allogenic responses to reconstructed human immune systems in a humanized mouse99. However, in human tumors, robust allogeneic responses are conferred by human T cells. Thus, the data collected from mouse models often do not precisely mirror the complex interactions between the tumor antigen-specific T cells and human tumor tissues in patients, thus offsetting the anticancer drug effects99. Therefore, nanoparticle distribution data in humanized mouse models where oncogenesis has taken place under the functional and autologous immune system will provide more predictive information regarding clinical outcomes.
Implications of using mouse-models to generate PK/PD data for nanoparticles
The bigger question that will eventually be required to answer is: what are the implications of such immunological difference between mice and humans in the context of nanoparticle treatment? This is a well-established fact that nanoparticles interact with different components of innate and adapted immunity post-administration. As discussed earlier, depending on design, composition, and chemistry, nanoparticles can have a varied impact on immune systems ranging from immunosuppression to immunomodulation. In turn, the contact of nanoparticles with circulating macrophages and reticuloendothelial systems (RES) affect the absorption, distribution, metabolism, and elimination (ADME) patterns of nanoparticles. Thus, if SPF-mice has immature immunity or 'neonate-like' immunity, nanoparticles' disposition pattern will be significantly different from that in any clinical subjects. Such non-representative disposition of nanoparticles in laboratory-raised mice will invariably lead to severely over-represented pharmacokinetic and pharmacodynamic activity of any drug molecules these nanoparticles can cargo within the mouse-model, which will have less realistic recapitulation when injected in humans. Age is another crucial factor that governs baseline immunity of laboratory mice, and in most in vivo experiments, mice of 6-8 weeks are used. However, age alone is a less predictable marker for the immune system's maturity. Comparison of feral and laboratory-raised mice clearly showed that laboratory mice have an immune system similar to neonatal humans, with lower innate immune activation and more naïve lymphocytes94.
On the contrary, mice subjected to diverse microbiological challenges have enhanced interferon and effector/memory lymphocytes, more like adult humans. Therefore, nanoparticle toxicity or efficacy data needs to be conducted on mice, closely matching human subjects' immune status. Such a proposition is not trivial so far as experimental reliability is concerned. In the next sections, we will discuss the current trends to generate more reliable data regarding immunological states of in vivo models for preclinical evaluation of nanoparticle efficacy.
Approaches to generate more reliable data from pre-clinical studies
The significant barrier to translating nanoparticle efficiency data from mouse models to humans is likely due to methodological discrepancies and the models' failure to recapitulate human disease conditions accurately. As indicated by Mak et al., unlike human clinical trials, no best practice standards are followed for in vivo, preclinical experiments100. Some of the methodological improvements proposed by this author include the use of both genders, varying age groups of mice, high-level randomization, outcomes assessor blinding, and publication of both negative and positive data. Approach-wise, the inclusion of higher animal species will result in more representative results. However, the experimental cost will produce severe challenges. Tian et al. has recently developed a humanized mouse model, known as Thy/HSC, that shows the spontaneous development of human B-ALL under autologous human immune surveillance. This was achieved by incorporating the leukemia-associated fusion gene, MLL-AF9, into human CD34+ FLCs, which were then co-transplanted with human fetal thymus tissue into NSG mice. Using this model, the group was able to show that recipient leukocyte infusions (RLI), a GVHD-free immunotherapeutic approach, significantly decreased human leukemia burden during induced lymphopenia101. This model was also used to study chimeric antigen T (CAR-T) cell therapy, for which a reduction of toxicity is still an unmet concern. When treated with anti-CD19 CAR-T cells, the Thy/HSC mouse model exhibited similar kinetics and levels to those observed in patients. In addition, rapid production of T cell- and myeloid cell-derived cytokines, such as GM-CSF, IFN-γ, TNF-α, and IL-10, and elevation of regulatory T cell frequency was also observed in these mice parallel to the observations reported in patients receiving CAR-T therapy102. Therefore, these humanized mouse models were a more reliable model to characterize human CAR-T cell function in vivo, leading to novel CAR therapies.
A novel humanized mouse model, named PDXv2.0, was recently constructed by the Nilsson group. In this model, in vitro expanded human tumor-infiltrating lymphocytes (TILs) were incorporated through adoptive transfer into the PDX mice that host the tumor collected from the same patients103. It was found that these PDX2.0 mouse model made by immunodeficient mouse recipients with human IL-2 continuous production efficiently represents the reactivity of adoptive cell transfer (ACT) immunotherapy that occurred in patients. Therefore, this model offered a reliable platform to model ACT-based immunotherapies and combinatory therapies for heterogeneous human cancers103.
As preceding sections indicate, SPF or laboratory-raised mice could significantly impact the immune-status of mice. Therefore, a critical re-evaluation of animal models and alternative strategies should be considered to promote smooth translation of nanoparticle efficiency data from bench to bedside. Mouse models are still the most popular and most prevalently used across laboratories world-wide to test the safety and efficacy of cancer therapeutics. Practical and ethical reasons have been paramount for such a versatile selection of mice as the most frequently used preclinical model. However, since this model is not sufficient to mirror the exact disposition of therapeutics in human subjects due to immune differences, the concept of 'microdoses' appears attractive. For example, in the last decade, both the FDA and the European Medicines Agency introduced guidelines for testing minimal doses of drugs in humans, comprising less than one-hundredth of the therapeutic dose104. Indicated as Phase 0 studies, a small number of patients are included for these studies to show how the therapeutic agent is distributed and metabolized in the body. Such experimentation will also bring about critical information regarding whether the drug is hitting its specific molecular target. Phase 0 trials, although very small in scope, will require a high-sensitivity detection mechanism. The approach to finding alternative mouse-model-based in vivo experimentation has also emerged, including epidemiological studies, autopsies, and in vitro studies using 'human organs on a chip' and Computer-based In silico modeling100. Machine-learning and systems biology have been proposed, and in many cases, tested to translate physiological and pathological relationships across species. A recent insight proposed by Brubaker and Lauffenburger proposed a newer approach to humanizing computational models derived from animal experiments rather than a humanizing approach. In contrast to cross-species pairs (CSP), which compare animal to human data set pairs, the authors proposed computational humanization, which shifts perspective from comparison to translating predictive models of biological associations across species, incorporating diverse molecular and phenotypic data from animals and humans105.
Outlook: Of all alternate approaches, it is safe to say that humanized rodents provide a much closer approximation of human physiology and pathology than any other models to predict nanoparticles' fate and disposition in the systemic circulation. Although the development of humanized mouse models with human immune systems and autologous human oncogenesis for more types of human tumors, including melanoma, lung cancer, hepatocarcinoma, and colorectal cancer, is still under development, pluripotent stem cell technology106 and gene editing tools, such as CRISPR/Cas9, may play crucial roles in spearheading these developments107. Using larger animal models, such as humanized pig models, are also attractive options in which nanoparticle kinetics could be studied at more physiologically relevant conditions. Studying nanoparticle distribution in immunodeficient mice with relevant human cytokine, chemokine, or ligand secretion under physiologically relevant conditions will promote more reliable data for matching preclinical with clinical data.
Future Perspectives
Several years of studies have witnessed the production of a vast array of nanomaterials derived from synthetic and natural sources. Several nanomaterials classes have also proved to be highly effective in combatting lethal diseases, especially cancers, at preclinical and even early-stage clinical trials. The most commonly faced hurdles in cancer therapy related to the drug's hydrophobicity, its rapid clearance, hepatic and renal toxicity, and off-target toxicity have been well addressed by using various nanoformulations. However, the transition from benchtop to bedside for a vast majority of the nanomaterials generated in laboratories world-wide is still a long way off.
The primary reason for this is attributed to insufficient knowledge of the nanosystems' functional aspects when in contact with biological components. The protein corona interaction still presents a challenge that slowly sheds light on the nanoparticles' fate when injected into the body. Another primary concern is the heterogeneity of cancer tissues, which vary with age, stage, gender, and race. Thus, a "one-size-fits-all" approach cannot be applied. The need for personalized nanomedicine based on the patient's clinical history and tumor type is becoming popular but is still in its early development stages. The ultimate fate of nanoparticles post-drug delivery also remains under-investigated. The advent of biomaterials and biocompatible polymers has reduced the risk of unwanted immune response and clearance to a large extent via modification of their size, composition, and targeting parameters. The future of nanomedicine looks bright, with a long path paved for humankind to reap its benefits.
Abbreviations
NPs, nanoparticles; PEG, poly(ethylene glycol), PLGA poly(lactic-co-glycolic acid); PLA, Polylactic acid; TCR, T-cell receptors; DC, dendritic cell; EPR, enhanced permeability and retention; SPR, surface plasmon resonance; CARPA, complement activation–related pseudoallergy; PAF, platelet-activating factor.
Author Contributions
Conceptualization, SKB, MQ, SB. Literature search, PR IH, NH, OM, SC, SB, SKB and MQ. Original draft, PR NH. Review and Editing, PR, IH, SB, SKB, MQ, AEB and MM Supervision, S.K.B.and Funding Acquisition, MQ. SKB, SB.
Acknowledgements
We thank the members of Midwest Veteran's biomedical Research Foundation (MVBRF) and KCMO research office. The work is supported by a Merit Review grant from the Department of Veterans Affairs (Sushanta K. Banerjee, 5I01BX001989-04 and Snigdha Banerjee, I01BX001002-05), KUMC Lied Basic Science Grant Program (SKB), and Grace Hortense Greenley Trust, directed by The Research Foundation in memory of Eva Lee Caldwell (SKB). Partial support for this work was received from NSF Grant No. IIA-1355466 from North Dakota Established Program to Stimulate Competitive Research (ND-EPSCoR) through the Center for Sustainable Materials Science and from NIH grant number 1 P20 GM109024 from the National Institute of General Medicine (NIGMS) (MQ).
Conflict of Interest
All authors declare no competing financial interests.
References
- Hammond PT. Nano Tools Pave the Way to New Solutions in Infectious Disease. ACS Infect Dis. 2017; 3(8): 554-558.doi:10.1021/acsinfecdis.7b00104.
- Poon Z, Chang D, Zhao X, et al. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano. 2011; 5(6): 4284-4292.doi:10.1021/nn200876f.
- Richards R, Bönnemann H. Synthesis Approaches to Metallic Nanomaterials, Apllications and Impact, WILEY-VCH. 2005:3
- Sekaly R-P. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? The Journal of experimental medicine. 2008; 205(1): 7-12.doi:10.1084/jem.20072681.
- Tomalia DA. In quest of a systematic framework for unifying and defining nanoscience. Journal of Nanoparticle Research. 2009; 11(6): 1251-1310.doi:10.1007/s11051-009-9632-z.
- Jain PK, El-Sayed MA. Noble metal nanoparticle pairs: effect of medium for enhanced nanosensing. Nano Lett. 2008;8(12):4347-4352.doi:10.1021/nl8021835.
- André E, Boutonnet B, Charles P, et al. A New, Simple and Versatile Strategy for the Synthesis of Short Segments of Zigzag-Type Carbon Nanotubes. Chemistry – A European Journal. 2016; 22(9): 3105-3114.doi:10.1002/chem.201503693.
- Guérineau V, Rollet M, Viel S, et al. The synthesis and characterization of giant Calixarenes. Nature Communications. 2019; 10(1): 113.doi:10.1038/s41467-018-07751-4.
- Clément M, Abdellah I, Ray P, et al. Synthesis and NMR study of trimethylphosphine gold(i)-appended calix[8]arenes as precursors of gold nanoparticles. Inorganic Chemistry Frontiers. 2020.doi:10.1039/C9QI01475F.
- Willis M, Forssen E. Ligand-targeted liposomes. Adv Drug Deliv Rev. 1998; 29(3): 249-271.doi:10.1016/s0169-409x(97)00083-5.
- Agut W, Brulet A, Schatz C, et al. pH and temperature responsive polymeric micelles and polymersomes by self-assembly of poly[2-(dimethylamino)ethyl methacrylate]-b-poly(glutamic acid) double hydrophilic block copolymers. Langmuir. 2010; 26(13): 10546-10554.doi:10.1021/la1005693.
- Mai Y, Eisenberg A. Self-assembly of block copolymers. Chem Soc Rev. 2012;41(18):5969-5985.doi:10.1039/c2cs35115c.
- Gillies ER, Fréchet JMJ. Dendrimers and dendritic polymers in drug delivery. Drug Discovery Today. 2005; 10(1): 35-43.doi:https://doi.org/10.1016/S1359-6446(04)03276-3.
- Wang L, Yin Y, Jain A, et al. Aqueous phase synthesis of highly luminescent, nitrogen-doped carbon dots and their application as bioimaging agents. Langmuir. 2014; 30(47): 14270-14275.doi:10.1021/la5031813.
- Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proceedings of the National Academy of Sciences. 2006; 103(16): 6315-6320
- Batist G, Barton J, Chaikin P, et al. Myocet (liposome-encapsulated doxorubicin citrate): a new approach in breast cancer therapy. Expert Opin Pharmacother. 2002; 3(12): 1739-1751.doi:10.1517/14656566.3.12.1739.
- Ermacora A, Michieli M, Pea F, et al. Liposome encapsulated daunorubicin (daunoxome) for acute leukemia. Haematologica. 2000; 85(3): 324-325
- Stone NRH, Bicanic T, Salim R, et al. Liposomal Amphotericin B (AmBisome(®)): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs. 2016; 76(4): 485-500.doi:10.1007/s40265-016-0538-7.
- Carnevale J, Ko AH. MM-398 (nanoliposomal irinotecan): emergence of a novel therapy for the treatment of advanced pancreatic cancer. Future Oncol. 2016; 12(4): 453-464.doi:10.2217/fon.15.333.
- Burade V, Bhowmick S, Maiti K, et al. Lipodox® (generic doxorubicin hydrochloride liposome injection): in vivo efficacy and bioequivalence versus Caelyx® (doxorubicin hydrochloride liposome injection) in human mammary carcinoma (MX-1) xenograft and syngeneic fibrosarcoma (WEHI 164) mouse models. BMC cancer. 2017; 17(1): 405-405.doi:10.1186/s12885-017-3377-3.
- Chaudhari MB, Desai PP, Patel PA, et al. Solid lipid nanoparticles of amphotericin B (AmbiOnp): in vitro and in vivo assessment towards safe and effective oral treatment module. Drug Deliv Transl Res. 2016; 6(4): 354-364.doi:10.1007/s13346-015-0267-6.
- Roy V, LaPlant BR, Gross GG, et al. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane®) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Annals of Oncology. 2009; 20(3): 449-453.doi:https://doi.org/10.1093/annonc/mdn661.
- Daull P, Lallemand F, Garrigue J-S. Benefits of cetalkonium chloride cationic oil-in-water nanoemulsions for topical ophthalmic drug delivery. The Journal of pharmacy and pharmacology. 2014; 66(4): 531-541.doi:10.1111/jphp.12075.
- Lallemand F, Schmitt M, Bourges J-L, et al. Cyclosporine A delivery to the eye: A comprehensive review of academic and industrial efforts. European Journal of Pharmaceutics and Biopharmaceutics. 2017; 117: 14-28.doi:https://doi.org/10.1016/j.ejpb.2017.03.006.
- Lombardo D, Kiselev MA, Caccamo MT. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. Journal of Nanomaterials. 2019; 2019: 3702518.doi:10.1155/2019/3702518.
- Sartor O. Eligard: leuprolide acetate in a novel sustained-release delivery system. Urology. 2003; 61(2 Suppl 1): 25-31.doi:10.1016/s0090-4295(02)02396-8.
- Pasut G. Pegylation of Biological Molecules and Potential Benefits: Pharmacological Properties of Certolizumab Pegol. BioDrugs. 2014; 28(1): 15-23.doi:10.1007/s40259-013-0064-z.
- Englert C, Brendel JC, Majdanski TC, et al. Pharmapolymers in the 21st century: Synthetic polymers in drug delivery applications. Progress in Polymer Science. 2018; 87: 107-164.doi:https://doi.org/10.1016/j.progpolymsci.2018.07.005.
- Evans ER, Bugga P, Asthana V, et al. Metallic nanoparticles for cancer immunotherapy. Materials Today. 2018; 21(6): 673-685.doi:https://doi.org/10.1016/j.mattod.2017.11.022.
- Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioengineering & translational medicine. 2019; 4(3): e10143-e10143.doi:10.1002/btm2.10143.
- Ko SH, Park I, Pan H, et al. Direct nanoimprinting of metal nanoparticles for nanoscale electronics fabrication. Nano letters. 2007; 7(7): 1869-1877
- Koseoglu-Imer DY, Kose B, Altinbas M, et al. The production of polysulfone (PS) membrane with silver nanoparticles (AgNP): physical properties, filtration performances, and biofouling resistances of membranes. Journal of membrane science. 2013; 428: 620-628
- Faraday M. The Bakerian Lecture: Experimental Relations of Gold (and Other Metals) to Light Philosophical Transactions of the Royal Society of London. 1857; 147: 145-181
- Webster TJ. Nanomedicine: what’s in a definition? International Journal of Nanomedicine. 2006; 1(2): 115-116
- Cuenca AG, Jiang H, Hochwald SN, et al. Emerging implications of nanotechnology on cancer diagnostics and therapeutics. Cancer. 2006; 107(3): 459-466.doi:10.1002/cncr.22035.
- Cliffel DE, Turner BN, Huffman BJ. Nanoparticle-based biologic mimetics. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009; 1(1): 47-59.doi:10.1002/wnan.20.
- Jain PK, Huang X, El-Sayed IH, et al. Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine. Acc Chem Res. 2008; 41(12): 1578-1586.doi:10.1021/ar7002804.
- Sharma A, Sharma US. Liposomes in drug delivery: progress and limitations. International journal of pharmaceutics. 1997; 154(2): 123-140
- Von Hoff DD, Mita MM, Ramanathan RK, et al. Phase I Study of PSMA-Targeted Docetaxel-Containing Nanoparticle BIND-014 in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016; 22(13): 3157-3163.doi:10.1158/1078-0432.CCR-15-2548.
- Sercombe L, Veerati T, Moheimani F, et al. Advances and Challenges of Liposome Assisted Drug Delivery. Frontiers in Pharmacology. 2015;6:286
- Ishida T, Harashima H, Kiwada H. Interactions of liposomes with cells in vitro and in vivo: opsonins and receptors. Curr Drug Metab. 2001; 2(4): 397-409.doi:10.2174/1389200013338306.
- Ulrich AS. Biophysical aspects of using liposomes as delivery vehicles. Biosci Rep. 2002; 22(2): 129-150.doi:10.1023/a:1020178304031.
- Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000; 156(4): 1363-1380.doi:10.1016/S0002-9440(10)65006-7.
- Soppimath KS, Aminabhavi TM, Kulkarni AR, et al. Biodegradable polymeric nanoparticles as drug delivery devices. Journal of Controlled Release. 2001; 70(1): 1-20.doi:https://doi.org/10.1016/S0168-3659(00)00339-4.
- Langer R. Biomaterials in drug delivery and tissue engineering: one laboratory's experience. Accounts of Chemical Research. 2000; 33(2): 94-101
- Fernández-Urrusuno R, Calvo P, Remuñán-López C, et al. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharmaceutical research. 1999; 16(10): 1576-1581
- Calderon M, Quadir M, Strumia M, et al. Functional dendritic polymer architectures as stimuli-responsive nanocarriers. Biochimie. 2010; 92(9): 1242-1251.doi:10.1016/j.biochi.2010.02.017.
- Bae Y, Kataoka K. Significant enhancement of antitumor activity and bioavailability of intracellular pH-sensitive polymeric micelles by folate conjugation. J Control Release. 2006; 116(2): e49-50.doi:10.1016/j.jconrel.2006.09.044.
- Ray P, Confeld M, Borowicz P, et al. PEG-b-poly (carbonate)-derived nanocarrier platform with pH-responsive properties for pancreatic cancer combination therapy. Colloids and Surfaces B: Biointerfaces. 2019; 174: 126-135.doi:https://doi.org/10.1016/j.colsurfb.2018.10.069.
- Ray P, Nair G, Ghosh A, et al. Microenvironment-sensing, nanocarrier-mediated delivery of combination chemotherapy for pancreatic cancer. J Cell Commun Signal. 2019; 13(3): 407-420.doi:10.1007/s12079-019-00514-w.
- Heinemann V. Gemcitabine: progress in the treatment of pancreatic cancer. Oncology. 2001; 60(1): 8-18.doi:10.1159/000055290.
- Pasut G, Canal F, Dalla Via L, et al. Antitumoral activity of PEG-gemcitabine prodrugs targeted by folic acid. Journal of controlled release : official journal of the Controlled Release Society. 2008; 127(3): 239-248.doi:10.1016/j.jconrel.2008.02.002.
- Abdullah CS, Ray P, Alam S, et al. Chemical Architecture of Block Copolymers Differentially Abrogate Cardiotoxicity and Maintain the Anticancer Efficacy of Doxorubicin. Molecular pharmaceutics. 2020.doi:10.1021/acs.molpharmaceut.0c00963.
- Karaca M, Dutta R, Ozsoy Y, et al. Micelle Mixtures for Coadministration of Gemcitabine and GDC-0449 To Treat Pancreatic Cancer. Molecular Pharmaceutics. 2016; 13(6): 1822-1832.doi:10.1021/acs.molpharmaceut.5b00971.
- Ray P, Ferraro M, Haag R, et al. Dendritic Polyglycerol-Derived Nano-Architectures as Delivery Platforms of Gemcitabine for Pancreatic Cancer. Macromol Biosci. 2019; 19(7): e1900073.doi: 10.1002/mabi.201900073.
- Calderón M, Quadir MA, Strumia M, et al. Functional dendritic polymer architectures as stimuli-responsive nanocarriers. Biochimie. 2010; 92(9): 1242-1251.doi: https://doi.org/10.1016/j.biochi.2010.02.017.
- Haag R, Kratz F. Polymer Therapeutics: Concepts and Applications. Angewandte Chemie International Edition. 2006; 45(8): 1198-1215.doi: 10.1002/anie.200502113.
- Confeld MI, Mamnoon B, Feng L, et al. Targeting the tumor core: hypoxia-responsive nanoparticles for delivery of chemotherapy to pancreatic tumors. Molecular Pharmaceutics. 2020.doi: 10.1021/acs.molpharmaceut.0c00247.
- Ray P, Kale N, Quadir M. New Side Chain Design for pH-Responsive Block Copolymers for Drug Delivery. Colloids and Surfaces B: Biointerfaces. 2021:111563.doi: https://doi.org/10.1016/j.colsurfb.2021.111563.
- Montet X, Funovics M, Montet-Abou K, et al. Multivalent effects of RGD peptides obtained by nanoparticle display. J Med Chem. 2006; 49(20): 6087-6093.doi: 10.1021/jm060515m.
- Rades N, Licha K, Haag R. Dendritic Polyglycerol Sulfate for Therapy and Diagnostics. Polymers. 2018; 10(6): 595.doi: 10.3390/polym10060595.
- Haag R. Supramolecular drug-delivery systems based on polymeric core-shell architectures. Angew Chem Int Ed Engl. 2004; 43(3): 278-282.doi: 10.1002/anie.200301694.
- Ray P, Alhalhooly L, Ghosh A, et al. Size-Transformable, Multifunctional Nanoparticles from Hyperbranched Polymers for Environment-Specific Therapeutic Delivery. ACS Biomaterials Science & Engineering. 2019; 5(3): 1354-1365.doi: 10.1021/acsbiomaterials.8b01608.
- Patri AK, Myc A, Beals J, et al. Synthesis and in vitro testing of J591 antibody-dendrimer conjugates for targeted prostate cancer therapy. Bioconjug Chem. 2004; 15(6): 1174-1181.doi: 10.1021/bc0499127.
- Ploegh HL. Logic of the immune system. Cancer Immunol Res. 2013; 1(1): 5-10.doi: 10.1158/2326-6066.CIR-13-0023.
- Corbo C, Molinaro R, Parodi A, et al. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine. 2015; 11(1): 81-100.doi: 10.2217/nnm.15.188.
- Kononenko V, Narat M, Drobne D. Nanoparticle interaction with the immune system. Arhiv za higijenu rada i toksikologiju. 2015; 66(2): 97-108.doi: 10.1515/aiht-2015-66-2582.
- Kim J-H, Kim C-S, Ignacio RMC, et al. Immunotoxicity of silicon dioxide nanoparticles with different sizes and electrostatic charge. Int J Nanomedicine. 2014; 9 183-193.doi: 10.2147/IJN.S57934.
- Alena Zhornik LB, Igor Volotovski, Sergey Chizhik, et al. Interaction of nanosilver particles with human lymphocyte cells. Adv Nat Sci: Nanosci Nanotechnol. 2015;6
- Chen L, Liu J, Zhang Y, et al. The toxicity of silica nanoparticles to the immune system. Nanomedicine. 2018; 13(15): 1939-1962.doi:10.2217/nnm-2018-0076.
- Zhao J, Stenzel MH. Entry of nanoparticles into cells: the importance of nanoparticle properties. Polymer Chemistry. 2018; 9(3): 259-272.doi: 10.1039/C7PY01603D.
- Ji Y, Wang N, Ramakrishnan R, et al. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem. 1999; 274(47): 33398-33402.doi: 10.1074/jbc.274.47.33398.
- Thuahnai ST, Lund-Katz S, Williams DL, et al. Scavenger receptor class B, type I-mediated uptake of various lipids into cells. Influence of the nature of the donor particle interaction with the receptor. J Biol Chem. 2001;276(47):43801-43808.doi: 10.1074/jbc.M106695200.
- Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014; 9 Suppl 1(Suppl 1): 51-63.doi: 10.2147/ijn.s26592.
- Kim S, Oh WK, Jeong YS, et al. Cytotoxicity of, and innate immune response to, size-controlled polypyrrole nanoparticles in mammalian cells. Biomaterials. 2011; 32(9): 2342-2350.doi: 10.1016/j.biomaterials.2010.11.080.
- Hirn S, Semmler-Behnke M, Schleh C, et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm. 2011; 77(3): 407-416.doi: 10.1016/j.ejpb.2010.12.029.
- Son YJ, Kim H, Leong KW, et al. Multifunctional nanorods serving as nanobridges to modulate T cell-mediated immunity. ACS nano. 2013; 7(11): 9771-9779.doi: 10.1021/nn403275p.
- Wibroe PP, Anselmo AC, Nilsson PH, et al. Bypassing adverse injection reactions to nanoparticles through shape modification and attachment to erythrocytes. Nat Nanotechnol. 2017; 12(6): 589-594.doi: 10.1038/nnano.2017.47.
- Liu Y, Yin Y, Wang L, et al. Surface hydrophobicity of microparticles modulates adjuvanticity. Journal of Materials Chemistry B. 2013; 1(32): 3888-3896.doi: 10.1039/C3TB20383B.
- Moyano DF, Saha K, Prakash G, et al. Fabrication of corona-free nanoparticles with tunable hydrophobicity. ACS Nano. 2014; 8(7): 6748-6755.doi: 10.1021/nn5006478.
- Chen F, Wang G, Griffin JI, et al. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat Nanotechnol. 2017; 12(4): 387-393.doi: 10.1038/nnano.2016.269.
- Walkey CD, Olsen JB, Song F, et al. Protein corona fingerprinting predicts the cellular interaction of gold and silver nanoparticles. ACS Nano. 2014; 8(3): 2439-2455.doi: 10.1021/nn406018q.
- Kwon YJ, Standley SM, Goh SL, et al. Enhanced antigen presentation and immunostimulation of dendritic cells using acid-degradable cationic nanoparticles. Journal of controlled release : official journal of the Controlled Release Society. 2005; 105(3): 199-212.doi: 10.1016/j.jconrel.2005.02.027.
- Liu X, Pu Y, Cron K, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015; 21(10): 1209-1215.doi: 10.1038/nm.3931.
- Foit L, Thaxton CS. Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials. 2016; 100: 67-75.doi: 10.1016/j.biomaterials.2016.05.021.
- Giovanni M, Yue J, Zhang L, et al. Pro-inflammatory responses of RAW264.7 macrophages when treated with ultralow concentrations of silver, titanium dioxide, and zinc oxide nanoparticles. J Hazard Mater. 2015; 297: 146-152.doi: 10.1016/j.jhazmat.2015.04.081.
- Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005; 216(2-3): 106-121.doi: 10.1016/j.tox.2005.07.023.
- Mazor R, King EM, Pastan I. Strategies to Reduce the Immunogenicity of Recombinant Immunotoxins. Am J Pathol. 2018; 188(8): 1736-1743.doi: 10.1016/j.ajpath.2018.04.016.
- Kishimoto TK, Ferrari JD, LaMothe RA, et al. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. 2016; 11(10): 890-899.doi: 10.1038/nnano.2016.135.
- Kim SH, Moon JH, Jeong SU, et al. Induction of antigen-specific immune tolerance using biodegradable nanoparticles containing antigen and dexamethasone. Int J Nanomedicine. 2019; 14: 5229-5242.doi: 10.2147/IJN.S210546.
- Freitag TL, Podojil JR, Pearson RM, et al. Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology. 2020; 158(6): 1667-1681 e1612.doi: 10.1053/j.gastro.2020.01.045.
- A G. The trouble with Animal Models. The Scientist. 2007
- Tao L, Reese T. Making Mouse Models That Reflect Human Immune Responses. Trends in Immunology. 2017; 38(3): 181-193.doi: 10.1016/j.it.2016.12.007.
- Reese T, Bi K, Kambal A, et al. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host & Microbe. 2016; 19(5): 713-719.doi: 10.1016/j.chom.2016.04.003.
- Beura LK, Hamilton SE, Bi K, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016; 532(7600): 512-516.doi: 10.1038/nature17655.
- Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity. 2009; 31(3): 368-376.doi: 10.1016/j.immuni.2009.08.009.
- Stelekati E, Shin H, Doering T, et al. Bystander Chronic Infection Negatively Impacts Development of CD8(+) T Cell Memory. Immunity. 2014; 40(5): 801-813.doi: 10.1016/j.immuni.2014.04.010.
- Stappenbeck T, Virgin H. Accounting for reciprocal host-microbiome interactions in experimental science. Nature. 2016; 534(7606): 191-199.doi: 10.1038/nature18285.
- De La Rochere P, Guil-Luna S, Decaudin D, et al. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018; 39(9): 748-763.doi: 10.1016/j.it.2018.07.001.
- Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014; 6(2): 114-118
- Xia J, Hu Z, Yoshihara S, et al. Modeling Human Leukemia Immunotherapy in Humanized Mice. EBioMedicine. 2016; 10: 101-108.doi: 10.1016/j.ebiom.2016.06.028.
- Jin CH, Xia J, Rafiq S, et al. Modeling anti-CD19 CAR T cell therapy in humanized mice with human immunity and autologous leukemia. EBioMedicine. 2019; 39: 173-181.doi: 10.1016/j.ebiom.2018.12.013.
- Jespersen H, Lindberg MF, Donia M, et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun. 2017; 8(1): 707.doi: 10.1038/s41467-017-00786-z.
- Marchetti S, Schellens J. The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. British Journal of Cancer. 2007; 97(5): 577-581.doi: 10.1038/sj.bjc.6603925.
- Brubaker DK, Lauffenburger DA. Translating preclinical models to humans. Science. 2020; 367(6479): 742.doi: 10.1126/science.aay8086.
- Rowe RG, Daley GQ. Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet. 2019; 20(7): 377-388.doi: 10.1038/s41576-019-0100-z.
- Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013; 154(6): 1380-1389.doi: 10.1016/j.cell.2013.08.021.