
The Role of TET Proteins in B Cell Biology
Shinya Tanaka1*, Wataru Ise2, Yoshihiro Baba1, Tomohiro Kurosaki2,3#
1Division of Immunology and Genome Biology, Medical Institute of Bioregulation, Kyushu University, Higashi-ku, Fukuoka, 812-0054, Japan
2Laboratory of Lymphocyte Differentiation, WPI Immunology Frontier Research Center, Osaka University, Osaka, Suita, 565-0871,Japan
3Laboratory of Lymphocyte Differentiation, RIKEN Center for Integrative Medical Sciences, Yokohama, Kanagawa, 230-0045, Japan
Abstract
Gene expression must be strictly controlled during cell differentiation and function in mammalian systems. DNA methylation plays an important role in this process, and its pattern is shaped by balancing the activity of methyltransferases and demethylases. Ten-eleven translocation (TET) was identified as a demethylase that catalyzes the oxidation reaction of the methyl group of 5-methylcytosine (5mC), converting it to 5-hydroxymethylcytosine (5hmC). Recently, indispensable roles of TET proteins in the regulation of immune cells have been identified. Here, we review recent studies on the biological consequences of dysregulation of TET proteins in the immune system, with a particular focus on B cell biology. Finally, we discuss future perspectives in this research field.
Introduction
The methylation pattern of eukaryotic DNA, which is critical for appropriate cell differentiation and function, is dynamically regulated by the activity of DNA methyltransferase and demethylases. Approximately ten years ago, Ten-eleven translocation 1 (TET1) was reported to act as a DNA demethylase in acute myeloid and lymphocytic leukemia1 and named after a t(10;11) (q22;q23) translocation. After this discovery, other TET family proteins TET2 and TET3 were identified by sequence homology. These TET proteins have a CpG DNA binding motif CXXC at the N-terminus (TET2 does not contain CXXC.) and a catalytic domain at the C-terminus2. In mammalian cells, these TET proteins catalyze the oxidation of 5-methylcytosine (5mC), sequentially generating 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), resulting in the generation of an unmethylated cytosine. Recent publications have reported roles of TET proteins other than their role as tumor suppressors3 in immune cells4-13. In this mini review, we discuss the role of TET proteins, especially in B cell biology.
Role of TET Proteins in Early B Cell Development
B cell-specific Tet2 and Tet3 gene-deficient mice using an Mb1Cre driver showed an impaired transition of Pro B to Pre B cells and defective Ig kappa (κ) gene rearrangement, which is a somatic rearrangement process of Vκ and Jκ gene segments to produce a functional Igκ light chain gene, during early B cell development5, 6. An analysis at the molecular level revealed that TET2/TET3 double-deficiency resulted in decreased Igκ locus germline transcripts and interferon regulatory factor (IRF)4/IRF8 expression5, 6, leading to speculation that TET-regulated IRF4/IRF8 might promote the expression of the germline transcripts, an activity that precedes and may be required for successful Ig light chain gene rearrangement. The Igκ region cis-regulatory elements, 3' enhancer (3'Eκ) and distal 3' enhancer (dEκ), contain CpG nucleotides, which are demethylated as early B cell development progresses5. In Pro B cells differentiated by co-culture with OP9 cells, disruption of the Tet2 and Tet3 genes enhanced DNA methylation at the 3'Eκ, which in turn reduced the amount of Ck transcripts and Igκ germline transcripts, accompanied by impaired chromatin accessibility of the Igκ locus. Therefore, DNA demethylation at 3’Ek is a crucial epigenetic event for Igκ gene rearrangement. Although overexpression of IRF4 did not restore the defective Igκ rearrangement caused by Tet deficiency, knock down of the E-protein E2A and the ETS-family protein PU.1 attenuated TET2-binding to Igκ cis-regulatory elements, with enhanced DNA methylation in Pre B cells, suggesting that E2A, PU.1 and TET coordinately regulate Igκ rearrangement and expression during early B cell development5.
Role of TET Proteins in Peripheral B Cell Differentiation
It has been reported that the TET proteins play a critical role in the differentiation of geminal center (GC) B and plasma cells11, 13, 14. Tet2/Tet3 deficiency did not affect cell proliferation, but formation of IgG1+ cells in vitro. A similar defect was observed in vivo after immunization 11, 13, even though the number of antigen-specific B cells was rather increased in the absence of TET2/TET3 in B cells11. Thereby, TET2/TET3 were suggested to play an important role in class switch recombination (CSR), which is an irreversible gene rearrangement process to generate different classes/isotypes of antibody. Consistent with this idea, the expression of activation-induced cytidine deaminase (AID), which is an essential mutagenic enzyme that catalyses the deamination of deoxycytidine to deoxyuracil to initiate CSR, somatic hypermutation and gene conversion, was substantially reduced in Tet2/Tet3-deficient B cells11. In fact, we have previously confirmed that acute Tet2/Tet3 deletion upon expression of a Tamoxifen-inducible Cre, decreased AID expression, accompanied by impairment of IgG1 CSR (unpublished data). IgG1 expression was restored in Tet2/Tet3-deficient B cells by enforced AID expression. On the other hand, the expression level of μ and γ1 germline transcripts in Tet2/Tet3-deficient B cells was equivalent to those of wild-type B cells. Therefore, the defective IgG1 CSR was solely due to decreased AID expression. Regarding the detailed molecular mechanism, it has already been reported that the transcription factor basic leucine zipper ATF-Like transcription factor (BATF) can induce AID expression by directly binding to the 5' region of the Aicda gene locus15. Lio et al. identified TET2-binding regions, named TetE1 and TetE2, in the 5' region of the Aicda locus and found that, in the absence of TET2/TET3, 5hmC modification was reduced at the TetE1, where BATF bound. In addition, TET2 and BATF were physically associated in primary B cells stimulated in vitro, suggesting that BATF and TET proteins coordinately induce AID expression11.
Role of TET Proteins in B Cell Tolerance
Self-tolerance is a vital biological event for homeostasis, as a break of self-tolerance is a direct cause of autoimmune disease. Peripheral B cell tolerance is established by death of self-reactive B cells, which is induced by discontinuous activation due to lack of T cell help. In other words, B cell peripheral tolerance is established by an intrinsic suppressive mechanism of self-reactive B cell activation and an extrinsic mechanism that prevents interaction of self-reactive B cells with self-reactive T cells. Although several endogenous factors that dampen self-reactive B cell activation have been identified18, little is known about the latter mechanism. In our current study, we discovered one of the mechanisms by which self-reactive T-B interaction is inhibited by TET2/TET3 to induce/maintain peripheral tolerance19.
In the above-cited publications5, 6, splenomegaly and lymphadenopathy were evident in Mb1 Cre x Tet2/Tet3 double floxed mice. We also found similar lymphoid tissue abnormalities in CD19 Cre-mediated Tet2/Tet3 conditional knock out mice (hereafter called Tet bDKO), but no conspicuous defects in early B cell development. As discussed in our publication19, this difference may be explained by the timing of Cre expression during early B cell development, since Mb1 Cre has been reported to be expressed earlier than CD1920, 21. Therefore, we hypothesized that the tissue abnormalities might have been caused by defective homeostasis of peripheral B cells, especially disruption of self-tolerance. In Tet bDKO mice, we found a constitutive activation of immune cells including B and T lymphocytes and myeloid cells, accompanied by autoimmune phenotypes: autoantibody production, cell infiltration into non-lymphoid tissues, and complement deposition in the kidneys. Consistent with these pathologies, Tet bDKO mice had renal damage19, suggesting development of a systemic lupus erythematosus (SLE)-like disease. Regarding the mechanism underlying autoimmune disease development in the Tet bDKO mice, introduction of a hen egg lysozyme (HEL)-specific B cell receptor (MD4) or B cell-specific MHC class II deletion into Tet bDKO mice suppressed the aberrant CD4+ T and B cell activation19. Therefore, antigen-mediated T-B interaction was required for the autoimmune reaction, suggesting that molecules which mediate T-B interaction may be responsible for dysregulated T and B cell activation. As might be expected, the gene encoding CD86, an essential co-stimulatory molecule for T cells, was identified by a comprehensive gene expression analysis done before disease onset among up-regulated differentially expressed genes in the absence of TET2/TET3 in B cells. On the other hand, there was almost no change in cytokine gene expression and in genes encoding other co-stimulatory molecules. Furthermore, a CD86 neutralizing experiment demonstrated that it was one of the responsible molecules for induction of spontaneous immune cell activation and autoimmune disease19.
Since it is difficult to identify self-reactive B cells in vivo, a transgenic model using HEL as a surrogate self-antigen is commonly used to study B cell tolerance22. In a peripheral tolerance model, self-reactive B cells are temporarily activated accompanied by CD86 induction upon HEL recognition, after which the activation is lost and the cells are eliminated by apoptotic cell death23. However, Tet2/Tet3-deficiency resulted in CD86 derepression and delayed elimination of HEL-specific B cells, suggesting that the absence of TET2/TET3 may increase the risk of self-reactive T cell activation, resulting in the development of autoimmune disease. Our findings are consistent with a previous report showing that enforced CD86 expression in self-reactive B cells could activate self-reactive T cells24. In addition, as only antigen stimulation without secondary co-stimulation makes self-reactive T cells anergic25, we could interpret these results to indicate that TET proteins expressed in B cells actively contribute to induction of peripheral T cell tolerance by CD86 suppression.
Based on these results, dysregulated CD86 was suggested to be one of the causes of the break in peripheral tolerance. However, the molecular mechanism by which CD86 expression was suppressed has been unknown. Although TET proteins were identified as DNA demethylases, they also function as adapters, recruiting histone deacetylase (HDAC) that contributes to the suppression of gene expression in the hematopoietic system through deacetylation4, 8. Therefore, we examined changes in DNA methylation, HDAC binding, and histone H3 acetylation caused by Tet2/Tet3 deletion in B cells at the genome-wide level. Our meta-analysis demonstrated that TET functioned both as a demethylase and an HDAC recruiter in B cells19. On the other hand, an integrated analysis of epigenetic data and gene expression data suggested that TET-dependent epigenetic changes optimize (fine-tune) transcription outcomes rather than being a determinant of whether genes are transcribed or not. At the Cd86 gene locus, HDAC bound in the promoter and 3’ region of Cd86 intron 1, and this binding was TET-dependent. Consistent with the reduced HDAC binding, the degree of acetylation of histone H3 in the promoter was enhanced19. Collectively, these results suggest that the TET proteins negatively regulate the expression of the Cd86 gene by forming HDAC-dependent suppressive chromatin structures. Although both TET-dependent HDAC binding and DNA demethylation were observed in close proximity to the 3’ region of Cd86 intron 1, it is currently unknown whether these two epigenetic events are functionally coordinated.
In our current study, we elucidated a TET-dependent inhibitory mechanism of T-B interaction that contributes to peripheral B cell tolerance. Previous studies demonstrated that CD86 expression was elevated in B cells from SLE and multiple sclerosis (MS) patients26, 27. In addition, a genome-wide association study (GWAS) identified TET2 and TET3 as risk factor genes in autoimmune diseases28, 29. Therefore, the mechanism of suppression of CD86 expression described above may be utilized to control autoimmunity in humans as well.
Conclusions and Future Perspectives
As described, TET2 and TET3 are required not only for B cell early development but also differentiation and homeostasis of mature peripheral B cells (Figure 1). However, despite the recent accumulating studies, the role of TET proteins in B cell biology is not yet fully understood. First, it has not been fully investigated exactly in which stages of B cell biology TET proteins are necessary. In particular, little research has been conducted to examine a role of TET in memory B cells. Second, it is still largely unclear at a molecular level how TET2 and TET3 regulate the above-mentioned key processes in B cell biology. For instance, recent reports suggest a critical role of TET2/TET3 in development and/or homeostasis of marginal zone (MZ) B cells and B1 cells, because these B cell subsets in spleen and peritoneal cavity almost completely disappeared in the absence of TET2/TET3 in B cells6, 19. However, the molecular mechanism underlying this phenotype is largely unknown. Third is the issue of independent roles of TET family proteins. In the current studies, doubly Tet2/Tet3 knock out cells have been utilized, because of the functional redundancy of TET2 and TET3. Therefore, it has been unclear whether there are specific genes independently regulated by each TET protein and if so, by what mechanism the selectivity is determined remains unclear. In addition, the role and downstream TET1 gene targets is largely undefined in the immune system. Regarding the more general question about TET’s molecular activities in the regulation of gene transcription beyond understanding of role of TET proteins in B cell biology, at least three questions should be answered: How many TET-target genes are regulated in either an HDAC-dependent or demethylation-dependent manner? In what context are these two molecular activities exerted? Do these two different TET activities coordinately regulate gene expression? A comprehensive understanding of these issues may be obtained by addressing them with various different cell types. Finally, as these basic research issues progress, whether the regulation of immune cells by manipulation of TET activity can serve as a novel approach to establish effective treatments for inflammatory diseases and cancer could be addressed.
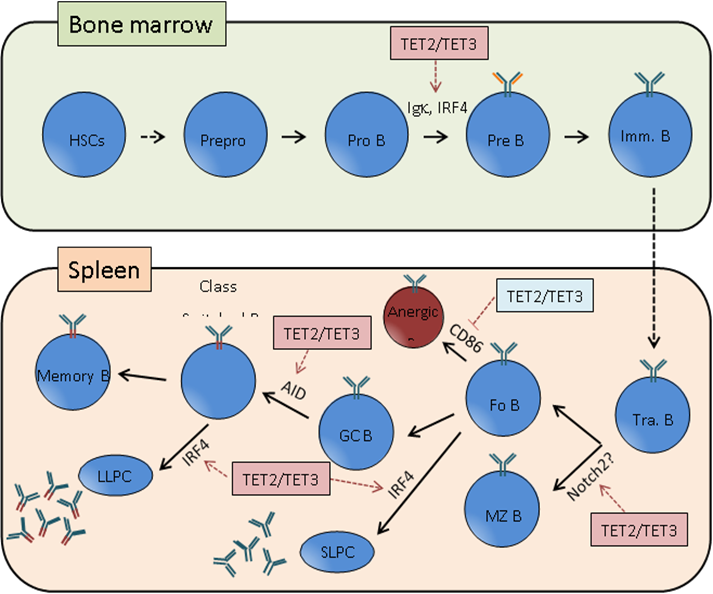
Figure 1: TET2/TET3 action points in B cell biology
Ten-eleven translocation (TET)2 and TET3 regulate key steps in B cell development and tolerance. The expression of Igκ, interferon regulatory factor (IRF) 4 and activation-induced deaminase (AID) are regulated by the demethylation activity of TET2/TET3. In contrast, CD86 expression is repressed by TET-dependent HDAC recruitment, mediating B cell peripheral tolerance.
Acknowledgement
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and AMED [Grant-in-Aid for Scientific Research (S) for T.K., AMED under grant no. 19gm6110004h0003 and Grant-in-Aid for Scientific Research (B) for Y.B and Grant-in-Aid for Scientific Research (C) for S.T.]
References
- Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930-935 (2009).
- Pastor W.A, et al. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14, 341-356 (2013).
- Cimmino L, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 16, 653-662 (2015).
- Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389-393 (2015).
- Lio C.W, et al. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 5 e18290 (2016).
- Orlanski S, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Natl Acad Sci U S A 113, 5018-5023 (2016).
- Montagner S, et al. TET2 Regulates Mast Cell Differentiation and Proliferation through Catalytic and Non-catalytic Activities. Cell Rep 15, 1566-1579 (2016).
- Xue S, et al. TET3 Inhibits Type I IFN Production Independent of DNA Demethylation. Cell Rep 16, 1096-1105 (2016).
- Yue X, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med 213, 377-397 (2016).
- Tsagaratou A, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol 18, 45-53 (2017).
- Lio C.J, et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol 4 eaau7523 (2019).
- Nakatsukasa H, et al. Loss of TET proteins in regulatory T cells promotes abnormal proliferation, Foxp3 destabilization and IL-17 expression. Int Immunol 31, 335-347 (2019).
- Schoeler K, et al. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. Febs J 286, 3566-3581 (2019).
- Fujii K, et al. Tet DNA demethylase is required for plasma cell differentiation by controlling expression levels of IRF4. Int Immunol 32, 683-690 (2020).
- Ise W, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol 12, 536-543 (2011).
- Ochiai K, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38, 918-929 (2013).
- Willis S.N, et al. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol 192, 3200-3206 (2014).
- Nemazee D, Mechanisms of central tolerance for B cells. Nat Rev Immunol 17, 281-294 (2017).
- Tanaka S, et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat Immunol 21, 950-961 (2020).
- Hobeika E, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A 103, 13789-13794 (2006).
- Audzevich T, et al. Pre/pro-B cells generate macrophage populations during homeostasis and inflammation. Proc Natl Acad Sci U S A 114, E3954-E3963 (2017).
- Goodnow C.C, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676-682 (1988).
- Cyster J.G, Goodnow C.C. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity 3, 691-701 (1995).
- Rathmell J.C, et al. Repression of B7.2 on self-reactive B cells is essential to prevent proliferation and allow Fas-mediated deletion by CD4(+) T cells. J Exp Med 188, 651-659 (1998).
- Schwartz R.H. T cell anergy. Annu Rev Immunol 21, 305-334 (2003).
- Smets I, et al. Multiple sclerosis risk variants alter expression of co-stimulatory genes in B cells. Brain 141, 786-796 (2018).
- Menard L.C, et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight 1, e87310 (2016).
- Yang W, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet 92, 41-51 (2013).
- Beecham A.H, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45, 1353-1360 (2013).