
Autophagy Modulation in Mammarenavirus Infection
Giovanna L. Gallo1*, Julieta S. Roldán2, Laura R. Delgui3,4
1Centro de Virología Animal (CEVAN)-CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas).
2Instituto de Investigaciones Biotecnológicas "Dr. Rodolfo A. Ugalde" (IIBIO), Universidad Nacional de San Martín (UNSAM) - CONICET, San Martín, Buenos Aires, Argentina.
3Instituto de Histología y Embriología de Mendoza (IHEM-CONICET). Facultad de Ciencias Médicas, Universidad Nacional de Cuyo, CONICET, Mendoza, Argentina.
4Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Cuyo, Mendoza, Argentina.
Abstract
Mammarenavirus genus groups viruses causing human haemorrhagic diseases, including the New World (NW) Junín virus (JUNV), and the Old World (OW) viruses Lassa (LASV), among others. The high mortality and morbidity rates associated to pathogenic mammarenaviruses, the absence of vaccines and the constant threat of new emerging species, make these viruses a public health concern in endemic areas. Autophagy is a widely-known intracellular metabolic pathway involved in maintaining the cellular homeostasis in response to several stress conditions.
In a previous work, we determined the role of autophagy in JUNV-infected cells. Our results indicate that JUNV, after being endocytosed and once the viral replication begins in the cytoplasm, induces an autophagic response in the infected cells and that a functional autophagy pathway is required for efficient virus replication. Recently, the interaction of OW LASV and the non- pathogenic one Mopeia (MOPV) with autophagic factors has been explored. The authors detected interaction with two well-known autophagic adaptors, NDP52 and TAX1BP1, and reported that, in Atg5 (an essential early autophagic protein) silenced permissive cells, less viral RNA and fewer infection particles were produced upon MOPV and LASV infection. Moreover, they observed that MOPV induced autophagy, which was required for efficient production of newly-formed infectious particles, suggesting a proviral role of autophagy. Here we discuss the implications of our findings in the context of others regarding mammarenavirus infection and the autophagy pathway.
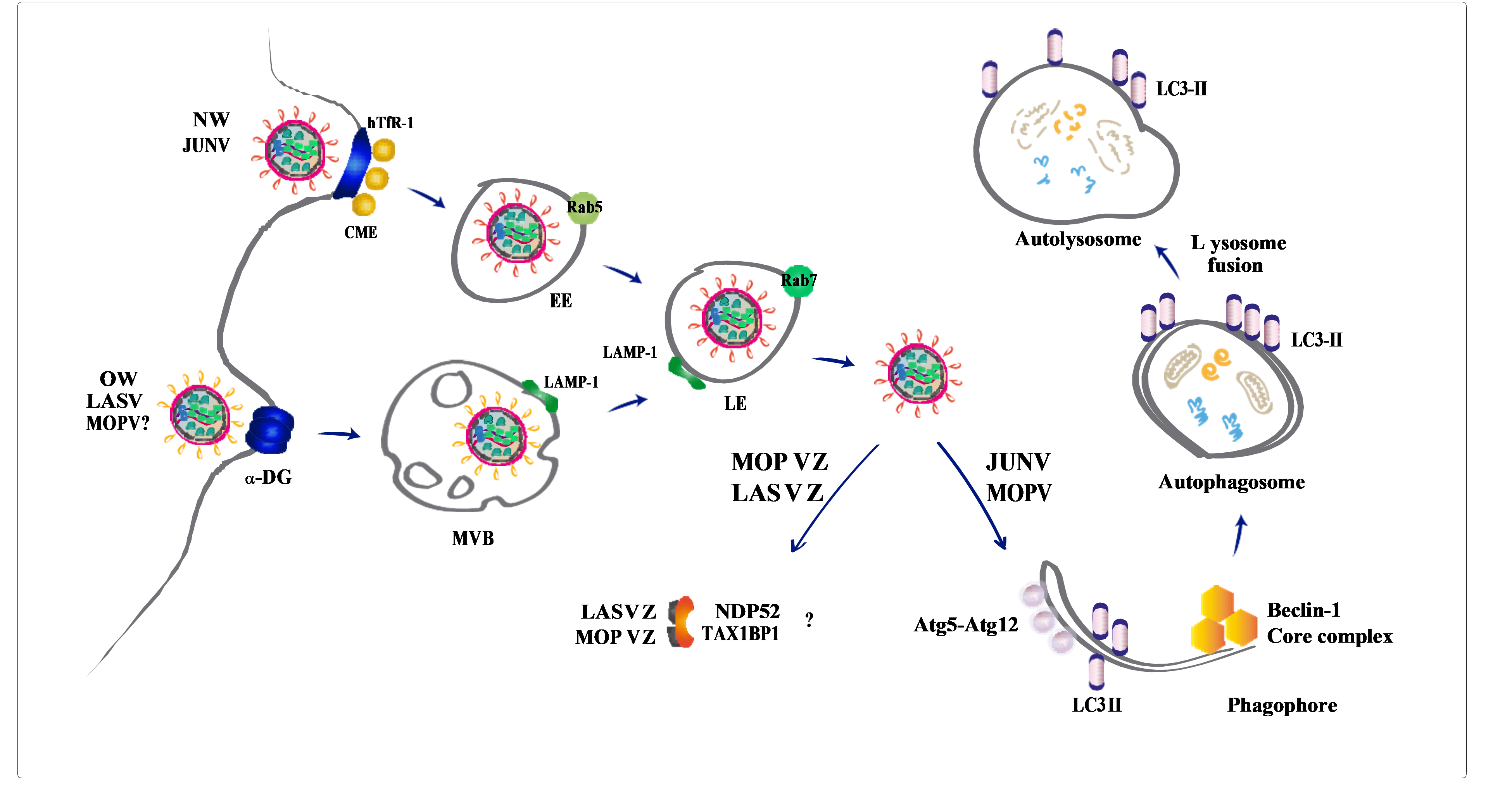
Figure 1.Graphical Abstract
Junín virus (JUNV), a member of Mammarenavirus genus within the Arenaviridae family, is the etiological agent of Argentine Haemorrhagic Fever (AHF), a potentially lethal, endemic-epidemic disease affecting the population of the most fertile farming land of Argentina1. Mammarenavirus genus groups viruses causing human HF diseases, including the New World (NW) viruses JUNV, Machupo, Guanarito, Sabia, and Chapare, and the Old World (OW) viruses Lassa (LASV) and the recently emerging Lujo virus. These two groups of mammarenavirus are divided based on phylogenetic, serological, and geographical distribution. Mammarenaviruses are negative-sense single strand RNA virus, with a bi-segmented genome consisting of a large (L) and small (S) molecule2. The L segment encodes the RNA-dependent RNA polymerase and the matrix protein (Z), a RING finger protein essential for viral morphogenesis and replication3,4. The S segment encodes the nucleoprotein (NP), responsible for nucleocapsid formation, and the Glycoprotein Precursor Complex (GPC). This polypeptide is exposed to proteolytic cleavage by cellular proteases into a stable signal peptide (SSP), GP1, and GP2 which remains associated and mediates recognition and entry into the target cell5. Mammarenaviruses represents a global threat with the emergence of new species as an increasingly possible event as the human population reaches new rural or previously inhabited territories. This aspect, together with the high mortality and morbidity rates associated to pathogenic mammarenaviruses and the absence of vaccines, makes these viruses a public health concern in endemic areas and led to their classification as category A pathogens by the U. S. National Institutes of Health (Emerging Infectious Diseases). To date, only Candid#1, a live-attenuated vaccine, is available in JUNV endemic areas from Argentina6.Autophagy is a widely-known intracellular metabolic pathway involved in maintaining the cellular homeostasis in response to several stress conditions7,8.However, autophagy also comprises an important activator of the innate and adaptive immunity9,10. Indeed, autophagy triggering is a commonly-used strategy by the cells to restrict viral infections, which can directly degrade the virus, and concomitantly regulate the innate and adaptive immunity to promote virus clearance11,12. This virus degradation leads to the activation of the Pattern Recognition Receptor signalling-cascade to finally induce type I interferon (IFN-I)-mediated viral elimination11,12. Thus, although the main function of autophagy during a viral infection is to counteract the infection, many viruses, most of them single-stranded RNA viruses, are able to hijack the autophagic pathway to facilitate viral replication, immune evasion and release13,14.
JUNV Promotes Autophagy
In our previous work, we analysed the autophagic response in JUNV infection by using the IV4454 JUNV strain15. We showed that JUNV triggers the accumulation of autophagic vesicles in a Beclin-1 and Atg5-dependent manner in permissive human A549 cells from 2 h post-infection (p.i.), indicating the early activation of the autophagic pathway after viral infection. Even though the precise role of autophagy during JUNV infection remains unclear to date, our results indicate that autophagy promotion has a proviral role toward JUNV replication, providing important knowledge to a previously unknown association in the field of host cell-arenavirus interactions15. Almost simultaneously, but using the more-virulent strain P3441 of JUNV as the viral model, the group of Perez Vidakovics also reported the autophagy-triggering by JUNV and both groups observed, in turn, that autophagy promotion enhances JUNV fitness in permissive A549 cells16. However, there were some differences in the results reported by both groups.
On one side, analysing early time-points after cell exposure, i.e. 2 and 6 h p.i., we observed that UV-inactivated JUNV infected cells were unable to promote autophagy, indicating that the autophagy activation was dependent on viral replication. Instead, Perez Vidakovics et al. showed, by western blot and fluorescence microscopy, an increased level of LC3-II lipidation (a well-established autophagosome indicator) after 24 h of cell exposure to UV-inactivated JUNV P3441 particles, suggesting that the autophagy activation was independent of viral replication. This discrepancy could be explained, in part, by the difference in the virulence between both strains used by us and Perez Vidakovics´s group. While we used the naturally-attenuated IV4454 JUNV strain (isolated from a mild human case of AHF in 197017), Perez Vidakovics et al. used the P3441 strain, isolated from another patient with AHF18. Certainly, virus virulence is determined by the virus–cell host net of interactions, so it becomes perfectly suitable to hypothesize that P3441 strain is able to induce autophagy by an alternative mechanism independently of viral replication, that is, by direct interaction of a structural component of the viral particle with the autophagy machinery once inside the cell. However, according to our results, GP signal was no longer observed 6 h p.i. by confocal laser scanning microscopy (CLSM), suggesting that the non-replicative inactivated-JUNV particles would be already degraded by 6 h p.i. This observation was not surprising to us since the UV light exposure generates RNA-protein cross-link that blocks genome transcription, eliminating any standard virus activity, leaving the particles to the mercy of cellular degradation mechanisms19,20. Indeed, it would be interesting to confirm the presence of intact inactivated viral particles after 24 h inside the cell, and dissect the mechanism that mediates the autophagy-induction by these particles.
Baird et al. found JUNV replication-transcription complexes (RTCs) associated with cellular membranes containing NP and mRNA , thus it was feasible to postulate that JUNV triggers autophagy to stimulate an intracellular membrane reprogramming that provides membrane scaffolds necessary to the formation of RTCs21. With this in mind, we analysed the possible co-localization of JUNV NP with the autophagic marker LC3 protein along the first 24 h of infection. While we did not detect co-localization either at early or late times p.i., i.e. 2 and 24 h p.i., respectively, Perez Vidakovics et al. observed co-localization of both structures at 24 h p.i. Regarding this bifurcated results in the distribution of viral and autophagic structures, in addition to the already-mentioned difference in strains virulence that could explain these observations, we found in methodological aspects the most feasible explanation. While we infected the cells with a multiplicity of infection (MOI) of 1, Perez Vidakovics et al. used a MOI of 3 and reported a Pearson’s coefficient of 0.71, so it could be possible that we missed the detection of NP-LC3 co-localization due to sensitivity issues within our approach. The authors suggested that the co-localization detected may implicate that the autophagosome membranes are involved in the RTCs assembly16. However, in the work by Baird and colleagues, they failed to detect co-localization of Candid#1 RTCs with LC3 by CLSM21. Indeed, further experiments are needed to elucidate the biological meaning of NP interaction with components of the autophagy machinery.
JUNV, Autophagy and ER Stress
Autophagy-triggering occurs by different stress signals to restore cellular homeostasis. Nutrient or growth factors deprivation, an excess of reactive oxygen species, aggregated or misfolded proteins, old or damaged organelles and several pathologic conditions are some of them7,22. A common effect of viral infections is the stress of the endoplasmic reticulum (ER), which is produced either by the exacerbated accumulation of viral proteins or the exploitation of ER membranes for viral replication10,23. To counteract the ER-stress, cells have evolved with the unfolded protein response (UPR)10,23,24. For JUNV, Paessler’s group recently showed that Candid#1 GPC (the protein that contains the most studied attenuation determinants of this strain) induces ER-stress promoting its degradation within lysosomal compartments25,26. They reported that a single amino acid substitution (T168A) in Candid#1 GPC resulted in the loss of an N-linked glycosylation motif and was the primarily responsible for the GPC retention in the ER, promoting the stress of the organelle. In our work, the JUNV-induced ER-stress was evaluated at early times p.i. by detecting the intracellular level of calnexin (CNX), a well-known ER-stress-induced protein27. However, we observed similar CNX levels in infected and non-infected cells, suggesting that the IV4454 JUNV strain triggers the autophagy pathway independently of the ER-stress. A possible explanation for these differences resides in the pathway of attenuation of both strains. While IV4454 is a naturally-attenuated strain, Candid#1 was attenuated after several animal passages and tissue culture cloning from the more pathogenic strain XJ 4428. In fact, we observed that IV4454 GPC does not possess the T168A substitution found in Candid#1.
Arenavirus and Autophagy: State of the Art
The Z protein is a small protein with key roles during the replication cycle thanks to its capacity of interaction with viral and cellular factors3,4. Hallam et al. reported recently that a single mutation in the RING domain of the Candid#1 Z protein is able to confer attenuation to the pathogenic Romero JUNV strain. This demonstrates that, besides the extensively studied GP protein, the Z protein is also an important determinant in Candid#1 strain attenuation25,29. Baillet et al. explored the interaction of the Z proteins from the highly pathogenic OW arenavirus LASV, and the non-pathogenic one, Mopeia (MOPV), with cellular factors using a human spleen cDNA library by yeast two-hybrid screening and coimmunoprecipitation. They detected interaction with two well-known autophagic adaptors, NDP52 and TAX1BP130. Also, they reported that, in Atg5 (an essential early autophagic protein) siRNA silenced HeLa cells, less viral RNA and fewer infection particles were produced upon MOPV and LASV infection. These results suggest that autophagy initiation structures, i. e. NDP52, TAX1BP1 and ATG5, play an important role in MOPV and LASV replication cycles30. Moreover, they observed that, 2 days after infection of permissive human HeLa cells, MOPV induced autophagy, which was required for efficient production of newly-formed infectious particles, suggesting a proviral role of autophagy. However, no autophagy-triggering was observed in LASV-infected cells. These results led the authors to suggest that autophagy may be required during different steps of MOPV and LASV replication cycles30.
A common observation between LASV, MOPV and JUNV with the autophagy machinery is their dependency on Atg5 expression for efficient virus replication15,16,30. Jounai et al. showed that autophagy may suppress the innate immune signalling via Atg5-Atg12 complex interaction with retinoic acid-inducible gene I pathway, leading to an impaired IFN-I response31. In their report, the authors observed an increase of IFN-I production in response to Vesicular Stomatitis Virus RNA in Atg5 deficient cells, inhibiting viral replication31. This IFN-I-inhibitory role of Atg5 suggests that autophagy may also have a regulatory role modulating the host innate antiviral response. In this context, one hypothesis is that the lower viral yield observed for JUNV, LASV and MOPV infection of Atg5 deficient cells, may be a consequence of enhanced IFN-I response rather than due to a deficiency in the autophagy flux. On the other side, it is well established that the NP protein of several mammarenaviruses acts as IFN-I antagonist in vitro32. So, putting all together and with the aim of depicting a unique scenario during JUNV infection in vitro, it becomes tempting to hypothesize that early in the infection (i.e. 2 h p.i.), the low levels of NP may not be sufficient to counteract the IFN-I response. Autophagy-triggering could comprise a viral strategy to promote an initial mechanism to down-regulate the IFN-I response. Then, at later times during the infection, an increased level of NP due to viral genome replication and protein translation could “take on the task” to down-regulate IFN-I antiviral action33,34. We have included all the discussed data and proposed hypothesis in Figure 2.
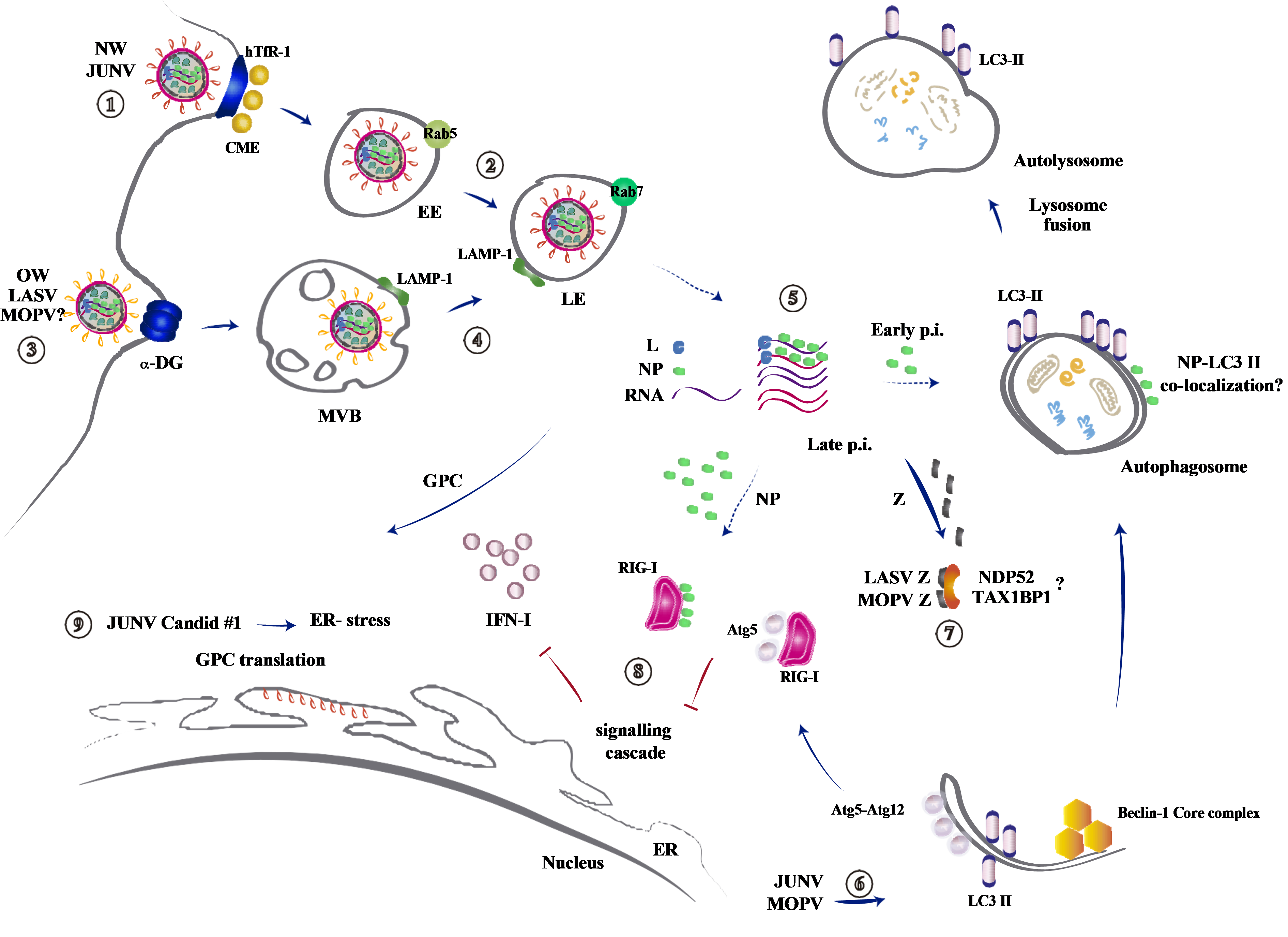
Figure 2.Interaction of mammarenaviruses with the autophagic pathway. 1) NW JUNV enters the cell by the human Transferrin Receptor 1 (hTfR)-136,37. Upon binding to hTfR-1, the virion is endocytosed by clathrin-mediated endocytosis (CME) and depends on the presence of Rab5 on the early endosomes (EE) for productive entry38,39. 2) The acidification of the late endosome (LE), characterized by the presence of Rab7, triggers the GPC-mediated fusion of viral and endosomal membranes and the release of the viral genome and replication complexes into the cell5,40-42. 3) OW LASV and MOPV viruses enter the cell by interaction with a-dystroglycan (a-DG) receptor, a ubiquitous and highly conserved cell-surface glycoprotein involved in adhesion to extracellular matrix43-45. 4) After internalization, the viruses transit through an unknown early endosomal compartment, possibly multivesicular bodies (MVB) and reach the LE. A pH-dependent receptor switch from a-DG to lysosome associated membrane protein (LAMP)-1 takes place (demonstrated for LASV46). Then, the acidic pH triggers the GPC-mediated fusion of viral and endosomal membrane42, 46-49. 5) By an unknown mechanism of “uncoating”, arenaviruses release their ribonucleoproteins comprised of viral RNA, the nucleocapsid protein (NP), and the viral polymerase (L) into the cytosol. Viral transcription initiates at the incoming polymerase complex, resulting in the early expression of NP and L50–52. Later times post-infection (p.i.), after genome replication, the glycoprotein complex (GPC) and Z proteins are translated53. 6-7) Arenaviruses interact with autophagy during the infectious cycle15,16,30. 6) JUNV induces the autophagic response in the infected cells and a functional autophagy pathway, requiring Atg5 and Beclin-1 proteins, is required for efficient virus replication. JUNV NP and LC3-II co-localization is not completely clear15,16. MOPV induces the autophagy pathway, but not LASV. 7) OW LASV and MOPV Z proteins, a late gene product in the viral replication cycle, interact with the autophagic adaptors NDP52 and TAX1BP1, and the complete infectious cycle requires the presence of Atg530. 8) Integrating the data, we propose that early p.i. the level of JUNV NP would be insufficient to reach its demonstrated type I interferon (IFN)-I antagonist role32-34. At that moment, the autophagy induction by the virus would offer augmented possibilities for Atg5 (from the Atg5-Atg12 complex) interaction with RIG-I to inhibit IFN-I response31. 9) GPC is translated in the endoplasmic reticulum (ER). JUNV Candid #1 GPC translation induces ER-stress, but not JUNV IV4454, thus JUNV IV4454-induced autophagy is due to an additional autophagy induction mechanism, unknown to date15,25. Solid and dotted-arrows indicate known and not completely known/proposed processes, respectively.
Conclusion
Taken all together, autophagy has a proviral role during JUNV, LASV and MOPV infection where the early structural autophagic protein Atg5 becomes crucial. To date, there is increasing evidence suggesting a proviral role of autophagy for several RNA viruses. For instance, foot-and-mouth disease virus triggers autophagy early times p.i. which is dependent on Atg5 but, contrary to what we observed for JUNV, independently of viral replication11,12,35. Some flaviviruses, including Dengue and Zika viruses, induce LC3-II-containing vesicles to regulate the storage of cellular lipids (a process known as lipophagy) and, in the case of Zika, promote the virus release11,12. Others, like Hepatitis C virus, its infection triggers autophagy through the UPR, which restrict IFN-I induction and so, promotes the viral replication 12,35. This ER-stress response could be similar to the one reported within Candid#1 infection but it seems not to be implicated during JUNV IV4454 autophagy induction15,26.
Regardless of slight differences among the observations, the evidences discussed in this work robustly demonstrate that JUNV triggers autophagy early after infection to establish an efficient viral infection in a human permissive cell line. Deeper exploration is needed to dissect if JUNV promotes autophagy to assist its replication, to suppress the innate immune signalling or a combination of both. All these results highlight the extensive interaction that might exist within mammarenaviruses and autophagy, and the different strategies developed by OW and NW arenaviruses in order to improve their fitness. Finally, and most importantly, these findings shed light on a new field of arenavirus host-cell interaction widening the panel of targets to develop new strategies to counteract these pathogens.
Acknowledgments
We sincerely thank Nélida A. Candurra for her enormous support and supervision during the elaboration of our original work, and Emily Bennison for the invitation to write this mini-review.
References
- Radoshitzky SR, Buchmeier MJ, Charrel RN, et al. ICTV virus taxonomy profile: Arenaviridae. J Gen Virol. 2019; 100(8): 1200-1201.
- Charrel RN, de Lamballerie X, Emonet S. Phylogeny of the genus Arenavirus. Curr Opin Microbiol. 2008; 11(4): 362-368. doi:10.1016/J.MIB.2008.06.001
- Loureiro ME, D’Antuono A, Levingston Macleod JM, et al. Uncovering viral protein-protein interactions and their role in arenavirus life cycle. Viruses. 2012; 4(9): 1651-1667. doi:10.3390/v4091651
- McLay L, Ansari A, Liang Y, et al. Targeting virulence mechanisms for the prevention and therapy of arenaviral hemorrhagic fever. Antiviral Res. 2013; 97(2): 81-92. doi:10.1016/j.antiviral.2012.12.003
- Nunberg JH, York J. The curious case of arenavirus entry, and its inhibition. Viruses. 2012; 4(1): 83-101.
- Ambrosio AM, Saavedra M del C, Riera LM, et al. La producción nacional de vacuna a virus Junin vivo atenuado (Candid #1) anti-fiebre hemorrágica argentina . Acta bioquímica clínica Latinoam. 2006; 40: 5-17.
- Ravanan P, Srikumar IF, Talwar P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017; 188: 53-67. doi:10.1016/j.lfs.2017.08.029
- Shah SWA, Chen D, Zhang J, et al. The effect of ammonia exposure on energy metabolism and mitochondrial dynamic proteins in chicken thymus: Through oxidative stress, apoptosis, and autophagy. Ecotoxicol Environ Saf. 2020; 206: 111413. doi:10.1016/j.ecoenv.2020.111413
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013; 13(10): 722-737. doi:10.1038/nri3532
- Cui J. Autophagy Regulation of Innate Immunity. Vol 1209. Springer Singapore; 2019. doi:10.1007/978-981-15-0606-2
- Mao J, Lin E, He L, et al. Autophagy and Viral Infection. Adv Exp Med Biol. 2019; 1209: 55-78. doi:10.1007/978-981-15-0606-2_5
- Choi Y, Bowman JW, Jung JU. Autophagy during viral infection—a double-edged sword. Nat Rev Microbiol. 2018; 16(6): 341-354.
- Lee YR, Lei HY, Liu MT, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008; 374(2): 240-248. doi:10.1016/J.VIROL.2008.02.016/li>
- Xiao Y, Cai W. Autophagy and Viral Infection. In: Autophagy: Biology and Diseases. Springer. 2020; 425-432.
- Roldán JS, Candurra NA, Colombo MI, et al. Junín Virus Promotes Autophagy To Facilitate the Virus Life Cycle. López S, ed. J Virol. 2019; 93(15). doi:10.1128/JVI.02307-18
- Perez Vidakovics MLA, Ure AE, Arrías PN, et al. Junín virus induces autophagy in human A549 cells. PLoS One. 2019; 14(6): 1-22. doi:10.1371/journal.pone.0218730
- Candurra NA, Damonte EB, Coto CE. Antigenic relationships between attenuated and pathogenic strains of Junin virus. J Med Virol. 1989; 27(2): 145-150.
- Gomez RM, Pozner RG, Lazzari MA, et al. Endothelial cell function alteration after Junin virus infection. Thromb Haemost. 2003; 90(08): 326-334.
- Ziegler CM, Eisenhauer P, Bruce EA, et al. The Lymphocytic Choriomeningitis Virus Matrix Protein PPXY Late Domain Drives the Production of Defective Interfering Particles. PLoS Pathog. 2016; 12(3): e1005501-e1005501. doi:10.1371/journal.ppat.1005501
- Damonte EB, Mersich SE, Coto CE. Response of cells persistently infected with arenaviruses to superinfection with homotypic and heterotypic viruses. Virology. 1983; 129(2): 474-478.
- Baird NL, York J, Nunberg JH. Arenavirus infection induces discrete cytosolic structures for RNA replication. J Virol. 2012; 86(20): 11301-11310. doi:10.1128/JVI.01635-12
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010; 40(2): 280-293. doi:10.1016/j.molcel.2010.09.023
- Jheng JR, Ho JY, Horng J. ER stress , autophagy , and RNA viruses. Front Microbiol. 2014; 5(August): 1-13. doi:10.3389/fmicb.2014.00388
- Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015; 40(3): 141-148. doi:10.1016/j.tibs.2015.01.002
- Seregin AV, Yun NE, Miller M, et al. The Glycoprotein Precursor Gene of Junin Virus Determines the Virulence of the Romero Strain and the Attenuation of the Candid #1 Strain in a Representative Animal Model of Argentine Hemorrhagic Fever. J Virol. 2015; 89(11): 5949-5956. doi:10.1128/jvi.00104-15
- Manning JT, Yun NE, Seregin AV, et al. The Glycoprotein of the Live-Attenuated Junin Virus Vaccine Strain Induces Endoplasmic Reticulum Stress and Forms Aggregates prior to Degradation in the Lysosome. J Virol. 2020; 94(8): 1-12. doi:10.1128/jvi.01693-19
- Ryan D, Carberry S, Murphy ÁC, et al. Calnexin, an ER stress-induced protein, is a prognostic marker and potential therapeutic target in colorectal cancer. J Transl Med. 2016; 14(1): 196. doi:10.1186/s12967-016-0948-z
- Ambrosio A, Saavedra M, Mariani M, et al. Argentine hemorrhagic fever vaccines. Hum Vaccin. 2011; 7(6): 694-700. doi:10.4161/hv.7.6.15198
- Hallam SJ, Manning JT, Maruyama J, et al. A single mutation (V64G) within the RING Domain of Z attenuates Junin virus. PLoS Negl Trop Dis. 2020; 14(9): e0008555. doi:10.1371/journal.pntd.0008555
- Baillet N, Krieger S, Journeaux A, et al. Autophagy Promotes Infectious Particle Production of Mopeia and Lassa Viruses. Viruses. 2019; 11(3): 293. doi:10.3390/v11030293
- Jounai N, Takeshita F, Kobiyama K, et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci. 2007; 104(35): 14050-14055.
- Martínez-Sobrido L, Giannakas P, Cubitt B, et al. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J Virol. 2007; 81(22): 12696-12703. doi:10.1128/JVI.00882-07
- Mateer EJ, Paessler S, Huang C. Visualization of Double-Stranded RNA Colocalizing With Pattern Recognition Receptors in Arenavirus Infected Cells. Front Cell Infect Microbiol. 2018; 8: 251. doi:10.3389/fcimb.2018.00251
- Huang C, Kolokoltsova OA, Yun NE, et al. Junín virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl Trop Dis. 2012; 6(5): e1659. doi:10.1371/journal.pntd.0001659
- Viret C, Rozières A, Faure M. Autophagy during Early Virus–Host Cell Interactions. J Mol Biol. Published online 2018. doi:https://doi.org/10.1016/j.jmb.2018.04.018
- Radoshitzky SR, Abraham J, Spiropoulou CF, et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007; 446(7131): 92-96. doi:10.1038/nature05539
- Helguera G, Jemielity S, Abraham J, et al. An Antibody Recognizing the Apical Domain of Human Transferrin Receptor 1 Efficiently Inhibits the Entry of All New World Hemorrhagic Fever Arenaviruses. J Virol. 2012; 86(7): 4024-4028. doi:10.1128/jvi.06397-11
- Martinez MG, Cordo SM, Candurra NA. Characterization of Junin arenavirus cell entry. J Gen Virol. 2007; 88(6): 1776-1784. doi:10.1099/vir.0.82808-0
- Martinez MG, Forlenza MB, Candurra NA. Involvement of cellular proteins in Junin arenavirus entry. Biotechnol J. 2009; 4(6): 866-870. doi:10.1002/biot.200800357
- Eschli B, Quirin K, Wepf A, et al. Identification of an N-Terminal Trimeric Coiled-Coil Core within Arenavirus Glycoprotein 2 Permits Assignment to Class I Viral Fusion Proteins. J Virol. 2006; 80(12): 5897-5907. doi:10.1128/jvi.00008-06
- Fedeli C, Moreno H, Kunz S. Novel Insights into Cell Entry of Emerging Human Pathogenic Arenaviruses. J Mol Biol. 2018; 430(13): 1839-1852. doi:10.1016/j.jmb.2018.04.026
- Rojek JM, Sanchez AB, Nguyen NT, et al. Different Mechanisms of Cell Entry by Human-Pathogenic Old World and New World Arenaviruses. J Virol. 2008; 82(15): 7677-7687. doi:10.1128/JVI.00560-08
- Cao W, Henry MD, Borrow P, et al. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998; 282(5396): 2079-2081. doi:10.1126/SCIENCE.282.5396.2079
- Spiropoulou CF, Kunz S, Rollin PE, et al. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J Virol. 2002; 76(10): 5140-5146. doi:10.1128/JVI.76.10.5140-5146.2002
- Reignier T, Oldenburg J, Noble B, et al. Receptor use by pathogenic arenaviruses. Virology. 2006; 353(1): 111-120. doi:10.1016/j.virol.2006.05.018
- Hulseberg CE, Fénéant L, SzymaÅska KM, et al. Lamp1 increases the efficiency of lassa virus infection by promoting fusion in less acidic endosomal compartments. MBio. 2018; 9(1): 1-14. doi:10.1128/mBio.01818-17
- Pasqual G, Rojek JM, Masin M, et al. Old World Arenaviruses Enter the Host Cell via the Multivesicular Body and Depend on the Endosomal Sorting Complex Required for Transport. Farzan M, ed. PLoS Pathog. 2011; 7(9): e1002232. doi:10.1371/journal.ppat.1002232
- Quirin K, Eschli B, Scheu I, et al. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology. 2008; 378(1): 21-33. doi:10.1016/j.virol.2008.04.046
- Rojek JM, Perez M, Kunz S. Cellular Entry of Lymphocytic Choriomeningitis Virus. J Virol. 2008; 82(3): 1505-1517. doi:10.1128/jvi.01331-07
- Lee KJ, Novella IS, Teng MN, et al. NP and L Proteins of Lymphocytic Choriomeningitis Virus (LCMV) Are Sufficient for Efficient Transcription and Replication of LCMV Genomic RNA Analogs. J Virol. 2000; 74(8): 3470-3477. doi:10.1128/jvi.74.8.3470-3477.2000
- López N, Jácamo R, Franze-Fernández MT. Transcription and RNA Replication of Tacaribe Virus Genome and Antigenome Analogs Require N and L Proteins: Z Protein Is an Inhibitor of These Processes. J Virol. 2001; 75(24): 12241-12251. doi:10.1128/jvi.75.24.12241-12251.2001
- Hass M, Gölnitz U, Müller S, et al. Replicon System for Lassa Virus. J Virol. 2004; 78(24): 13793-13803. doi:10.1128/jvi.78.24.13793-13803.2004
- Hallam SJ, Koma T, Maruyama J, et al. Review of mammarenavirus biology and replication. Front Microbiol. 2018; 9(AUG). doi:10.3389/fmicb.2018.01751