
Expansion of Gamma Delta T Cells - A Short Review on Bisphosphonate and K562-Based Methods
Wee Kiat Tan1, Johan CK Tay2, Jieming Zeng3, Min Zheng4, Shu Wang2,3*
1Tessa Therapeutics, Pte Ltd., Singapore 239351
2Department of Biological Sciences, National University of Singapore, Singapore 117543
3Institute of Bioengineering and Nanotechnology, Singapore 138669
4Department of Dermatology, Second Affiliated Hospital, Zhejiang University, School of Medicine, China 310009
Gamma Delta T cells As the Next Generation of Effector Cells for Immunotherapy
Gamma delta (γδ)-T cells are thymus-derived lymphocytes that differ from αβ-T cells in their anatomical distributions and mechanisms of activation and function. They could be found as resident lymphocytes in human tissues or as circulating lymphocytes in peripheral blood1 (Vantourout & Hayday, 2013). Unlike αβ-T cells that function exclusively in adaptive immunity, γδ-T cells are innate-like immune cells that recognize malignant cells through their repertoire of activating receptors in a MHC-independent manner, which is similar to natural killer cells2 (Welsh et al., 1997).
Growing interest in the use of γδ-T cells as an alternative form of immunotherapy has been fueled partially by the MHC-restriction mechanism that limits αβ-T cell-based cancer immunotherapy to an autologous setting. γδ-T cells recognize antigens independently of MHC molecules and this mode of action avoids GvHD when used in an allogenic setting3,4 (Deniger et al., 2014; Xiao et al., 2018). Owing to their repertoire of activating receptors, γδ-T cells have been reported to kill a wide variety of cancer cell lines in vitro such as leukemia, melanoma, lymphomas and other carcinomas5 (Yoshikawa et al., 2014). Drawing inspiration from the growing field of chimeric antigen receptors (CAR), mRNA CAR-engrafted αβ-T and γδ-T cells have been investigated in parallel and were found to exhibit comparable cytolytic activity against melanoma cells. However, γδ-T cells produced less proinflammatory cytokines as compared with αβ-T which make patients infused with γδ-T cells less susceptible to cytokine release syndrome6 (Harrer et al., 2017). Given the above qualities, γδ T cells may be produced in large quantity as an off-the-shelf agent for cancer immunotherapy.
γδ-T cells consist of 2 major subtypes, namely Vδ1 TCR and Vδ2 TCR subtype. Both subtypes could be found in the peripheral blood but the Vδ1 subtype is predominantly found in the epithelial tissues. The Vδ1 subtype reacts strongly to stress-associated factors commonly presented on cancer cells and virus-infected cells and act to destroy these cells, hence forming the first line of defense in the epithelial tissues7 (Ebert, Meuter, & Moser, 2006). (Vantourout & Hayday, 2013) 1Furthermore, Vδ1 γδ-T cells have been found to directly kill Gram-positive bacteria found in the gut and promote wound healing through enhancing keratinocyte proliferation on the skin8 (Nielsen, Witherden, & Havran, 2017).
In the peripheral blood, γδ-T cells only compose 0.5 to 5% of peripheral blood mononuclear cells (PBMCs) whereas αβ-T cells make up approximately 50%. Among these circulating γδ-T cells, Vγ9Vδ2 T cells are the majority subset that has been well-studied for their anti-tumour activity9 (Davodeau et al., 1993) but only account for less than 10% of all T cells10 (Dopfer et al., 2014). The infrequent nature of γδ-T cells had therefore impeded their clinical application. However, the fortuitous finding in the 1990s that biophosphonates could activate the in vivo proliferation of Vγ9Vδ2 T cells during treatment of bone resorption pioneered the search for more expansion methods. On this basis, derivative methods such as cytokine-based and K562 feeder cell-based protocols have been developed to achieve clinically relevant numbers for cancer immunotherapy. In this review, we survey the current landscape of γδ-T cell culturing methods that address the need in Vγ9Vδ2-based cancer immunotherapy.
Bisphosphonate-Based Expansion of Vγ9Vδ2 T Cells
Cytokine-based approaches for expansion of γδ-T cells have thus far revolved around the combinatorial use of both IL-2 and Zoledronate (ZOL) or the synthetic phosphoantigen bromohydrin pyrophosphate (BrHPP). ZOL and BrHPP share structural homology with non-peptide γδ-T cell ligands such as the isopentenyl pyrophosphate (IPP) intermediate found in the mevalonate pathway. The farnesyl pyrophosphate synthase (FPPS) enzyme in the mevalonate pathway converts IPP to farnesyl pyrophosphate, and the various metabolites generated through this pathway have been implicated in tumour growth and progression. Nitrogen-containing bisphosphonates (N-BPs) such as ZOL inhibit the FPPS enzyme through competitive inhibition11 (Kavanagh et al., 2006) and though unclear, synthetic analogues such as BrHPP could be functioning in a similar fashion.
Addition of ZOL or BrHPP to PBMC culture blocks FPPS activity and causes an accumulation of IPP in monocytes12 (Roelofs et al., 2009) and MHC Class II+ cells13 (Soriano-Sarabia et al., 2012), which collectively activates Vγ9Vδ2 T cells through cell-to-cell interactions. Although Vγ9Vδ2 T cells can function as antigen-presenting cells (APCs) to CD8+ cytotoxic T lymphocytes (CTLs)5 (Yoshikawa et al., 2014), it is unclear if they cross-present IPP for mutual activation. In earlier publications, Vγ9Vδ2 T cells were purified from PBMCs before culturing with FPPS inhibitors and IL-2. As described by Salot et al., labelled Vγ9Vδ2 T cells were sorted from PBMCs with magnetic beads; the unlabeled cell fraction was irradiated and used for co-culturing with the sorted Vγ9Vδ2 T cells14 (Salot et al., 2009). Although gamma irradiation may reduce concomitant expansion of non-γδ T cells, antigen presenting capabilities of professional APCs could also be impaired by such treatment15 (Merrick et al., 2005) which may explain the low expansion folds of Vγ9Vδ2 T cells using this method.
Besides APCs, CD4 T cells and their secreted cytokines also contribute to the activation of Vγ9Vδ2 T cells. In a Mycobacterium tuberculosis model, Vγ9Vδ2 T cells were primed by cytokines secreted by CD4 T cells such as IFNγ and IL-216 (Pechhold, Wesch, Schondelmaier, & Kabelitz, 1994). The removal of CD4+ and CD8+ T cells may, therefore, have contributed to lowered expansion fold of the Vγ9Vδ2 T cells17 (Dokouhaki et al., 2010) although this could be partially rescued by exogenous IL-2.
In these cytokine-based approaches, the length of culture was often kept between 10 – 14 days (Table 1). Vγ9Vδ2 T cells harvested from day 12 to day 14 might be most optimal for therapy as cytotoxic activity peaked on day 12 and declined by day 1418 (Kondo et al., 2008). Prolonged exposure of Vγ9Vδ2 T cells to phosphoantigens beyond 14 days caused activation-induced cell death (AICD)19 (Ferrarini et al., 2008). Therefore, lengthening the culture period in pursuit of increased Vγ9Vδ2 T cell expansion runs the risk of producing exhausted Vγ9Vδ2 T cells with lower cytotoxic activity.
Although the highest rate of cytokine-based expansion reported by Kondo et al. stands at 13750 fold20 (Kondo et al., 2011), all other methods reported a range of between 180 to 5000-fold (Table 1). Therefore, the infrequent nature of Vγ9Vδ2 T cells necessitates a large starting number of PBMC to generate adequate effector Vγ9Vδ2 T cells for ACT. Estimating at an average therapeutic dose of 2x106 γδ-T cells/kg bodyweight21 (Wilhelm et al., 2014) and an average human body weight of 62kg22 (Walpole et al., 2012), a typical ACT would require at least 1.2x108 cells per infusion. This means that leukapheresis, which is an invasive and strenuous procedure in order to obtain adequate starting PBMCs, becomes inevitable and hence complicating the process of Vγ9Vδ2 T cell generation. In order to develop an off-the-shelf cancer immunotherapy, a larger expansion rate of the Vγ9Vδ2 T cell is needed to streamline the process and lower the cost of production.
Table 1: Comparison of bisphosphonate and cytokine based methods for γδ-T cell expansion
| Cytokines and Reagents Used | Duration (Days) | γδ-T Expansion Fold | Nature of Sorting Procedure Performed | Reference |
|---|---|---|---|---|
| IFNγ, IL-12, anti-CD2 mAb, anti-CD3 mAb (OKT3), IL-2 | 14 | Not determined; 70-fold change for total cell count | After initial expansion | (Liu, Guo, Gehrs, Nan, & Lopez, 2005)34 |
| BrHPP, IL-2, OKT3 | 14 | 1585 ± 1493 | No | (Salot et al., 2007)35 |
| ZOL, IL-2 | 14 | 4798 ± 654 | No | (Kondo et al., 2008)18 |
| BrHPP, IL-2, OKT3 | 10 | 180 | PBMCs were sorted with MACS “TCR γ/δ + T cell isolation kit” before coculture with irradiated remainder of PBMCs | (Salot et al., 2009)14 |
| IL-2, IL-4, OKT3 | 10 to 14 | 376 ± 185 | CD4 and CD8 T cell depletion with RosetteSep depletion kit before culture | (Dokouhaki et al., 2010)17 |
| ZOL, IL-2, IL-18 | 14 | 1000-5000 | N.D | (Li et al., 2010)36 |
| ZOL, IL-2 | 10 to 14 | 13750 | No | (Kondo et al., 2011)20 |
| ZOL, IL-2, IL-15 | 14 | 1000 | No | (Van Acker et al., 2016)37 |
Feeder Cell-Based Vγ9Vδ2 T Cells Expansion Methods for Clinical Use
The various reports reviewed thus far have highlighted a need to conserve the non-γδ T cell fraction during activation by Zol and IL-2, and a need to consider the duration of culture such that a clinically-sufficient number of Vγ9Vδ2 T cell could be obtained for adoptive cell therapy.
To generate even larger numbers of effector cells, artificial antigen-presenting cells (aAPCs) have been explored as a feeder cell-based method to provide a more sustained source of activation and costimulation. K562 is an MHC-negative human chronic erythroleukemic cell line that has been genetically modified and adapted for cell-based expansion of polyclonal CTLs23 (Maus et al., 2002) and natural killer cells24 (Imai, Iwamoto, & Campana, 2005). K562 was initially chosen as a cellular scaffold for genetic modification because the lack of MHC class I and class II expression prevents allogeneic responses from donor T lymphocytes23 (Maus et al., 2002).
Deniger and colleagues were the first group to adopt this aAPC technology for the expansion of polyclonal γδ-T cells3 (Deniger et al., 2014) (Table 2). After the use of microbeads to remove NK cells and positively-select for polyclonal γδ-T cells, the authors used K562 feeder cells, modified to express CD19, CD64, CD86, CD137L and membrane-bound IL15, to expand and maintain the polyclonal γδ-T populations. These K562 cells were able to induce an expansion of 4900 ± 1700 folds (Table 2), which is a clear improvement from figures obtained from cytokine-based methods (Table 1). Unlike two later papers that focused on the Vγ9Vδ2 subset25,4 (Cho et al., 2016; Xiao et al., 2018), Deniger used these feeder cells to expand and study the polyclonal population and also the isolated Vδ1 and Vδ2 subsets, and interestingly found that the previously less-studied Vδ1 subset was capable of elucidating anti-tumour effects as well. In addition, the use of K562 cells as aAPC seemed to negate the AICD effects of prolonged exposure to antigens. Over a 35-day co-culture period, Deniger reported up to 107-fold increase in umbilical cord blood (UCB)–derived γδ-T cells3 (Deniger et al., 2014). The authors opined that this application is potentially useful as the younger UCB-derived γδ-T cells contain a more diverse repertoire of TCRγδ and would support a longer-term engraftment in allogeneic recipient patients.
Table 2: Comparison of K562-based γδ-T cell expansion methods
| Cytokines and Reagents Used | γδ-T Expansion Fold | Co-Culture Ratio of T Cell to Feeder Cell | Genetic Modification of K562 Feeder Cells | Frequency of Stimulation with Feeder Cells | Nature of Sorting Performed | Reference |
|---|---|---|---|---|---|---|
| IL-2, IL-21 | 4900±1700 | 1 to 2 | CD19, CD64, CD86, CD137L, membrane-bound IL-15 | every 7 days | CD56 microbeads for removal of NK cells; TCRγ/δ+ T cell isolation kit for positive selection of γδ-T cells | (Deniger et al., 2014)3 |
| IL-2, αCD3, αCD28 | 12-fold at day 14, 1,000,000-fold by day 100 | 2 to 1 | CD32, CD80, CD137L | every 7 days | FACS-based positive selection of Vγ9Vδ2 T cells prior to co-culture with aAPCs | (Cho et al., 2016)25 |
| ZOL, IL-2, OKT3 | 537340±607389 | 1 to 100 | CD64, CD86, CD137L | once on day 7 | Magnetic bead-based removal of αβ-T cells from day 7 culture before stage 2 culture with K562 aAPCs | (Xiao et al., 2018)4 |
Also working on Vγ9Vδ2 T cells, Cho and colleagues used a different K562 cell line that was modified to express CD32, CD83 and CD137L instead to expand Vγ9Vδ2 T cells purified from PBMCs by FACS sorting. Unlike the expansion figures achieved by Deniger et al., Cho and colleagues were only able to achieve an approximately 12-fold expansion by day 14 and a 106-fold by day 10025 (Cho et al., 2016). In terms of clinical applications, this adds significantly to the manufacturing costs and duration.
Concerns with the use of K562 feeder cells for immune cell expansion lie primarily with the potential presence of residual K562 cells after co-culture and post-infusion into patients. To address this, K562 cells are gamma-irradiated to arrest their cell growth and to prevent unintended in vivo proliferation after ACT. Various studies, including ours, have reported very low or undetectable levels of gamma-irradiated K562 feeder cells after 7 days of co-culture26,4 (Lapteva et al., 2012; Xiao et al., 2018). FDA regulations require that there should be less than 0.1% of detectable gamma-irradiated feeder cells in the final product26 (Lapteva et al., 2012). As such, immune effector cells expanded by K562 feeder cells have been approved for clinical trials by the FDA (NKEXP and NKCD19; clinicaltrials.gov numbers NCT00640796 and NCT00995137).
Combinatorial Usage of ZOL, OKT3 and K562 Feeder Cells in an FDA-Registered G-Rex Device
In our work, we had chosen to focus on the Vγ9Vδ2 T cell population because it is the subset that has been most extensively studied for its safety and efficacy27,28,29 (Fisher, Heuijerjans, Yan, Gustafsson, & Anderson, 2014; Fournie et al., 2013; Kobayashi & Tanaka, 2015). This sets our work apart from that performed by Deniger although their expansion protocol for a polyclonal γδ-T cell population remains impressive. The key factor that differentiates our work from Cho et al. then is the initial stage of enriching Vγ9Vδ2 T cell from the general PBMC population by ZOL and IL-2 as compared to cell sorting. As explained in the previous section, the use of ZOL to enrich Vγ9Vδ2 T cell has been well-validated and requires the presence of accessory cells such as monocytes and other MHC class II-positive cells. The use of K562 feeder cells, as demonstrated by Deniger et al., does not seem to favour any particular γδ-T subset and merely serves to numerically expand the population. The two-stage protocol developed in our paper, therefore, leverages on the ability of ZOL to enrich Vγ9Vδ2 T cells, and the ability of K562 feeder cells to numerically expand the enriched population.
In our two-stage protocol (Figure 1), a robust and selective activation of Vγ9Vδ2 T cells was achieved in the first phase through the use of ZOL prior to sorting4 (Xiao et al., 2018). A well-timed magnetic sorting at day 7 after activation would remove NK and αβ T cell contaminants but maintain the activation status of Vγ9Vδ2 T cells. This is evident in the potent fold expansion of Vγ9Vδ2 T cells of 537340 ± 607389 in 17 days. This is the fastest and highest Vγ9Vδ2 T cell expansion figure we have observed to date. Although there may be concern arising from the high ratio of Vγ9Vδ2 T to K562 used at 1:100, we could prove minimal residual feeder cells at day 17, and that the overall usage of K562 feeder cells was limited to once for the whole duration.
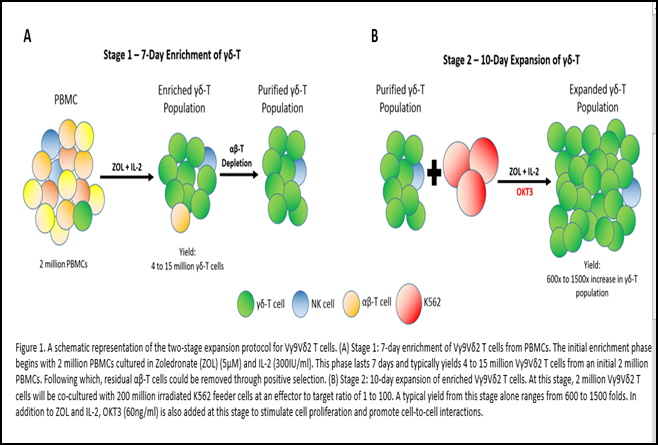
The fact that K562 cells could support a longer-term in vitro culture, as compared to the typical 10-14 day period in cytokine-based methods, was also demonstrated in our own study4 (Xiao et al., 2018). We had reported that the 10-day co-culture period with K562 brought about a decrease in classic T cell exhaustion markers such as CTLA-4, LAG3, TIM3 and BTLA4. Day-17 Vγ9Vδ2 T cells were activated and highly cytotoxic against ZOL-treated cancer cells. Cytotoxicity assays also did not reveal any difference between day 7 ZOL/IL-2-expanded Vγ9Vδ2 T cells and day 17 K562-expanded Vγ9Vδ2 T cells (unpublished data). Although there was no observable in vitro difference in cytotoxicity, the down-regulated expression of immune checkpoint inhibitors could possibly nullify the strongly immunosuppressive tumour microenvironment and maintain in vivo anti-tumour activity.
The huge numerical expansion is explained by the usage of anti-CD3 antibody OKT3 that targets the CD3 epsilon chain expressed on γδ-T cells. Importantly, we built on the interesting observation by Dopfer et al. that OKT3 could promote significant cytokine secretion and proliferation by Vγ9Vδ2 T cells10 (Dopfer et al., 2014). Our K562 feeder cell line is engineered to express CD64, which is a high-affinity human IgG FcγRI receptor. The interaction between OKT3 and CD64 thus promotes cell-to-cell contact between K562 and Vγ9Vδ2 T cells, which in turn enables better interactions between CD86 and CD137L on expressed on our K562 feeder cell line and the CD28 and CD137 costimulatory molecules expressed on Vγ9Vδ2 T cells respectively.
To set the stage for future clinical trials and applications with these K562-expanded Vγ9Vδ2 T cells, we trialed the use of the G-Rex (Wilson Wolf, US) system in the second stage of our expansion protocol. G-Rex is designed to support cell cultures with low cell seeding density at 0.125x106/cm2 30(Bajgain et al., 2014). Its key feature is the gas-permeable silicone membrane at the bottom that facilitates efficient exchange of O2 and CO2 which enables more media to be added per unit of surface area. By doing so, the frequency of media exchange is reduced, and the increased nutrient support also enhances cell survival31 (Vera et al., 2010). Its compact design and lower requirement for manpower also enable scalability in large-scale cell manufacturing which is an important consideration for commercialization. In our work, we seeded 1x106 day 7 ZOL-activated Vγ9Vδ2 T cells with 1x108 irradiated-K562 feeder cells at a coculture ratio of 1:1004 (Xiao et al., 2018). With this FDA-registered class I medical device, we could achieve an expansion of between 1,000 to 15,000 folds in just 10 days. The use of G-Rex thus enables us to achieve a clinically-relevant number of cells needed for effective therapeutic intervention.
The Role of Vγ9Vδ2 T Cells in Th1 Cytokine Secretion and Chimeric Antigen Receptor
Recently, there has been emerging concern about the pathogenic nature of certain γδ-T cells. Indeed, it has been noted that polyclonal γδ-T cells expanded by K562 aAPC secrete Th17 cytokines including IL-17 when activated by a leukocyte activation cocktail3 (Deniger et al., 2014). Yet, in the same experiment, it was shown that there was a significant secretion of interferon-gamma (IFN-γ), which is a good surrogate marker for cytotoxicity. Additional experiments had shown that the Vδ2 subset was the main source of IFN-γ, indicating that Vγ9Vδ2 T cells might be the majority population of the polyclonal γδ-T cells. Another review pointed out that γδ-T cells may present Th2 or Th17 profile based on the cytokine milieu within the microenvironment. Yet, these findings are largely based on results from in vitro experiments and not pre-clinical models32 (Xiang & Tu, 2017). This remains a concern and more preclinical data should be collected to understand the mechanism behind γδ-T cells adopting a Th17 profile in certain tumour microenvironment. On our part, we have mitigated this risk by focusing on the Vγ9Vδ2 subset, which is known to express a Th1 signature with the majority producing proinflammatory IFN-γ and a minority (<5%) secreting IL-1733 (Patil, Bhat, Dar, & Chiplunkar, 2015).
The combined use of ZOL and Vγ9Vδ2 T cells presents clear therapeutic effect in clinical trials with patients achieving partial remission in advanced breast and prostate cancer and complete remission of RCC patient with lung metastasis. However, certain cancer cells are resistant to ZOL treatment and do not respond well to Vγ9Vδ2 T cells. To enhance the anti-tumour activity of these K562-expanded Vγ9Vδ2 T cells, we have shown in our work that CAR-engrafted Vγ9Vδ2 T cells function efficiently against EpCAM-positive cancer cells. Given their propensity to home in to epithelial tissues and the antigen-specificity that could be afforded by CAR technology, γδ-T cells have huge potential for clinical applications against various solid tumours.
Conclusion and Future Perspective
A significant milestone in gene therapy and adoptive cell therapy was achieved with the recent FDA approval of CAR-T cells for use in relapsed B-cell malignancies. However, the dependence on autologous αβ-T cells to drive this anti-tumour effort has placed a huge constraint on logistics and supply chain management, from individualized sample collection from patient, to the ex vivo expansion, genetic modification and quality control. This has considerably driven up costs and time needed for effective therapeutic interventions. In this aspect, Vγ9Vδ2 T cells represent a hugely viable option to replace conventional αβ-T cells as an off-the-shelf immunotherapeutic agent. The intrinsic anti-tumour activity of γδ cells, coupled with their MHC-independent modus operandi, makes them ideal allogenic off-the-shelf effector cells for cancer immunotherapy. The bottleneck had been the large-scale expansion of these scarce cells, and this has been overcome to various extents by the various methods reported. We believe that future clinical trials involving Vγ9Vδ2 T cells would explore the combinatorial use with other therapeutic agents, such as CAR and immune checkpoint inhibitors. With our recent publication4 (Xiao et al., 2018), we have provided an economical method to achieve large-scale expansion of these Vγ9Vδ2 T cells and have also validated their combinatorial use with CAR technology. Our use of CD64, CD86 and CD137L-expressing K562 feeder cells has also mitigated the risks of having exhausted Vγ9Vδ2 T cells from an extended period of in vitro culture. Future efforts in our laboratory and others should strive to constantly improve the quality of the expanded Vγ9Vδ2 T cells while reducing the cost of therapy, thereby allowing the therapy to reach the masses efficiently.
References
- Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013; 13(2): 88-100. doi:10.1038/nri3384
- Welsh RM, Lin MY, Lohman BL, et al. Alpha beta and gamma delta T-cell networks and their roles in natural resistance to viral infections. Immunol Rev. 1997; 159: 79-93.
- Deniger DC, Maiti SN, Mi T, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin Cancer Res. 2014; 20(22): 5708-5719. doi:10.1158/1078-0432.CCR-13-3451
- Xiao L, Chen C, Li Z, et al. Large-scale expansion of Vgamma9Vdelta2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy. 2018; 20(3): 420-435. doi:10.1016/j.jcyt.2017.12.014
- Yoshikawa T, Takahara M, Tomiyama M, et al. Large-scale expansion of gammadelta T cells and peptide-specific cytotoxic T cells using zoledronate for adoptive immunotherapy. Int J Oncol. 2014; 45(5): 1847-1856. doi:10.3892/ijo.2014.2634
- Harrer DC, Simon B, Fujii SI, et al. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer. 2017; 17(1): 551. doi:10.1186/s12885-017-3539-3
- Ebert LM, Meuter S, Moser B. Homing and Function of Human Skin T Cells and NK Cells: Relevance for Tumor Surveillance. The Journal of Immunology. 2006; 176(7): 4331-4336. doi:10.4049/jimmunol.176.7.4331
- Nielsen MM, Witherden DA, Havran WL. gammadelta T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. 2017; 17(12): 733-745. doi:10.1038/nri.2017.101
- Davodeau F, Peyrat MA, Hallet MM, et al. Peripheral selection of antigen receptor junctional features in a major human gamma delta subset. Eur J Immunol. 1993; 23(4): 804-808. doi:10.1002/eji.1830230405
- Dopfer EP, Hartl FA, Oberg HH, et al. The CD3 conformational change in the gammadelta T cell receptor is not triggered by antigens but can be enforced to enhance tumor killing. Cell Rep. 2014; 7(5): 1704-1715. doi:10.1016/j.celrep.2014.04.049
- Kavanagh KL, Guo K, Dunford JE, et al. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci U S A. 2006; 103(20): 7829-7834. doi:10.1073/pnas.0601643103
- Roelofs AJ, Jauhiainen M, Monkkonen H, et al. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br J Haematol. 2009; 144(2): 245-250. doi:10.1111/j.1365-2141.2008.07435.x
- Soriano-Sarabia N, Sandvold H, Jomaa H, et al. Primary MHC-class II(+) cells are necessary to promote resting Vdelta2 cell expansion in response to (E)-4-hydroxy-3-methyl-but-2-enyl-pyrophosphate and isopentenyl pyrophosphate. J Immunol. 2012; 189(11): 5212-5222. doi:10.4049/jimmunol.1200093
- Salot S, Bercegeay S, Dreno B, et al. Large scale expansion of Vgamma9Vdelta2 T lymphocytes from human peripheral blood mononuclear cells after a positive selection using MACS "TCR gamma/delta+ T cell isolation kit". J Immunol Methods. 2009; 347(1-2): 12-18. doi:10.1016/j.jim.2009.05.006
- Merrick A, Errington F, Milward K, et al. Immunosuppressive effects of radiation on human dendritic cells: reduced IL-12 production on activation and impairment of naive T-cell priming. Br J Cancer. 2005; 92(8): 1450-1458. doi:10.1038/sj.bjc.6602518
- Pechhold K, Wesch D, Schondelmaier S, et al. Primary activation of V gamma 9-expressing gamma delta T cells by Mycobacterium tuberculosis. Requirement for Th1-type CD4 T cell help and inhibition by IL-10. J Immunol. 1994; 152(10): 4984-4992.
- Dokouhaki P, Han M, Joe B, et al. Adoptive immunotherapy of cancer using ex vivo expanded human gamma delta T cells: A new approach. Cancer Letters. 2010; 297(1): 126-136. doi:10.1016/j.canlet.2010.05.005
- Kondo M, Sakuta K, Noguchi A, et al. Zoledronate facilitates large-scale ex vivo expansion of functional gammadelta T cells from cancer patients for use in adoptive immunotherapy. Cytotherapy. 2008; 10(8): 842-856. doi:10.1080/14653240802419328
- Ferrarini M, Delfanti F, Gianolini M, et al. NF-kappa B modulates sensitivity to apoptosis, proinflammatory and migratory potential in short- versus long-term cultured human gamma delta lymphocytes. J Immunol. 2008; 181(9): 5857-5864.
- Kondo M, Izumi T, Fujieda N, et al. Expansion of human peripheral blood gammadelta T cells using zoledronate. J Vis Exp. 2011; (55). doi:10.3791/3182
- 21. Wilhelm M, Smetak M, Schaefer-Eckart K, et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. Journal of Translational Medicine. 2014; 12(1): 45. doi:10.1186/1479-5876-12-45
- Walpole SC, Prieto-Merino D, Edwards P, et al. The weight of nations: an estimation of adult human biomass. BMC Public Health. 2012; 12(1): 439. doi:10.1186/1471-2458-12-439
- Maus MV, Thomas AK, Leonard DG, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol. 2002; 20(2): 143-148. doi:10.1038/nbt0202-143
- Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005; 106(1): 376-383. doi:10.1182/blood-2004-12-4797
- Cho HW, Kim SY, Sohn DH, et al. Triple costimulation via CD80, 4-1BB, and CD83 ligand elicits the long-term growth of Vgamma9Vdelta2 T cells in low levels of IL-2. J Leukoc Biol. 2016; 99(4): 521-529. doi:10.1189/jlb.1HI0814-409RR
- Lapteva N, Durett AG, Sun J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. 2012; 14(9): 1131-1143. doi:10.3109/14653249.2012.700767
- Fisher JP, Heuijerjans J, Yan M, et al. gammadelta T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology. 2014; 3(1): e27572. doi:10.4161/onci.27572
- Fournie JJ, Sicard H, Poupot M, et al. What lessons can be learned from gammadelta T cell-based cancer immunotherapy trials. Cell Mol Immunol. 2013; 10(1): 35-41. doi:10.1038/cmi.2012.39
- Kobayashi H, Tanaka Y. gammadelta T Cell Immunotherapy-A Review. Pharmaceuticals (Basel). 2015; 8(1): 40-61. doi:10.3390/ph8010040
- Bajgain P, Mucharla R, Wilson J, et al. Optimizing the production of suspension cells using the G-Rex "M" series. Mol Ther Methods Clin Dev. 2014; 1: 14015. doi:10.1038/mtm.2014.15
- Vera JF, Brenner LJ, Gerdemann U, et al. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex). J Immunother. 2010; 33(3): 305-315. doi:10.1097/CJI.0b013e3181c0c3cb
- Xiang Z, Tu W. Dual Face of Vγ9Vδ2-T Cells in Tumor Immunology: Anti- versus Pro-Tumoral Activities. Frontiers in Immunology. 2017; 8: 1041. doi:10.3389/fimmu.2017.01041
- Patil RS, Bhat SA, Dar AA, et al. The Jekyll and Hyde story of IL17-Producing gammadeltaT Cells. Front Immunol. 2015; 6: 37. doi:10.3389/fimmu.2015.00037
- Liu Z, Guo BL, Gehrs BC, et al. Ex vivo expanded human Vgamma9Vdelta2+ gammadelta-T cells mediate innate antitumor activity against human prostate cancer cells in vitro. J Urol. 2005; 173(5): 1552-1556.
- Salot S, Laplace C, Saiagh S, et al. Large scale expansion of gamma 9 delta 2 T lymphocytes: Innacell gamma delta cell therapy product. J Immunol Methods. 2007; 326(1-2): 63-75. doi:10.1016/j.jim.2007.07.010
- Li W, Kubo S, Okuda A, et al. Effect of IL-18 on expansion of gammadelta T cells stimulated by zoledronate and IL-2. J Immunother. 2010; 33(3): 287-296. doi:10.1097/CJI.0b013e3181c80ffa
- Van Acker HH, Anguille S, Willemen Y, et al. Interleukin-15 enhances the proliferation, stimulatory phenotype, and antitumor effector functions of human gamma delta T cells. J Hematol Oncol. 2016; 9(1): 101. doi:10.1186/s13045-016-0329-3