
RIC-3, a potential target for regulating cholinergic signaling and inflammation
Millet Treinin*
Abstract
The nicotinic acetylcholine receptor (nAChR) gene family encodes for subunits of acetylcholine gated ion channels. These receptors are expressed widely and have many functions including anti-inflammatory effects mediated by the α7 nAChR, as part of the cholinergic anti-inflammatory pathway, in immune cells, microglia and astrocytes. Maturation of α7 nAChRs into functional ligand-gated ion channels in the plasma membrane is a complex process likely to require the RIC-3 protein. This endoplasmic reticulum resident chaperone affects maturation of multiple nAChRs, but its interaction with these receptors and its effects on their maturation differ for different nAChRs. Moreover, these interactions and effects are regulated by multiple mechanisms. Genetic analysis has implicated RIC-3 in the neuroinflammatory disease Multiple Sclerosis (MS), and in the neurodegenerative Parkinson's disease (PD). Neuroinflammation contributes to the progression of neurodegenerative diseases including PD. This information combines to suggest that RIC-3 may contribute to progression of both MS and PD via its effects on the α7 nAChR and the cholinergic anti-inflammatory pathway. Furthermore, we suggest that mechanisms regulating RIC-3 expression and activity may have a role in controlling inflammation.
The nicotinic acetylcholine receptor family
The nicotinic acetylcholine receptors (nAChRs) are a large and diverse family of acetylcholine-gated ion channels. In mammals this family is composed of nine alpha subunits and seven non-alpha subunits. These subunits assemble to form mostly heteromeric (α7 and α9 nAChR subunits can form homomeric receptors), cation-selective channels having diverse properties and expression patterns. In skeletal-muscles and in the autonomic nervous system nAChRs mediate excitatory synaptic transmission. However, in the central nervous system (CNS) nAChRs do not usually mediate synaptic transmission and instead play modulatory roles, including a role in regulating neurotransmitter release, reviewed by Dani and Bertrand1. In addition to being expressed in muscles and neurons, nAChRs are also found in multiple types of non-excitable cells where they affect migration, differentiation, proliferation, and signal transduction2.
CNS-expressed nAChRs have been implicated in memory, cognition, addiction, and several neurodegenerative diseases. Evidence for these functions comes from the addictive effects of tobacco-derived nicotine and from epidemiological studies linking tobacco smoking to neurodegenerative diseases such as schizophrenia and Parkinson's disease (reviewed by Dani and Bertrand1). In addition, decreased brain expression of nAChRs was shown in Alzheimer’s disease, Parkinson's disease (PD), Lewy-body dementia, and schizophrenia3-5.
Alpha7 nAChR and the cholinergic anti-inflammatory pathway
The α7 nAChR was first identified as the α-bungarotoxin binding receptor in the CNS6. The wide distribution and high calcium permeability of this receptor suggested an important role for this nAChR7,8. However, knockout mice are viable and show no gross abnormalities in development or behavioral phenotypes9. In contrast, in humans, copy number variations in the gene encoding for the α7 subunit, CHRNA7, are associated with brain diseases such as epilepsy or autism10. Understanding the role of CHRNA7 in diseases of the nervous system is, unfortunately, hampered by the fact that most deletions encompassing this gene also cover additional genes. Nevertheless, behavioral phenotypes associated with these deletions are likely to be a result of CHRNA7 haploinsufficiency as rare small heterozygote deletions covering CHRNA7 alone are associated with similar phenotypes to those associated with larger deletions10. This conclusion, however, is complicated by findings showing that some heterozygous carriers of CHRNA7 containing deletions are phenotypically normal, possibly a result of incomplete penetrance of these deletions10.
CHRNA7 knockout mice show no readily discernable phenotypes9. Nevertheless, α7 nAChR has one well-established role, i.e., mediating the anti-inflammatory effects of acetylcholine, as part of the cholinergic anti-inflammatory pathway. This pathway enables inhibition of inflammation by vagal efferents and depends on α7 nAChR expression in immune cells11,12. For its anti-inflammatory effects, α7 nAChR functions via activation of signaling cascades such as MAPK and NF-kappaB and via regulation of gene expression13-15. This pathway, first shown to function in macrophages, is now known to function in multiple immune cells and in glial cells, micrglia, and astrocytes, having immune functions in the CNS, reviewed by Treinin et. al.16.
Maturation of nAChRs and the RIC-3 protein
Maturation of nAChRs is a complex and inefficient process requiring assistance from cellular factors. Several proteins were shown to affect functional expression and properties of nAChRs. These proteins differ in specificity and mode of action, as reviewed by Treinin17. The best characterized of these proteins are LYNX1, functioning at the plasma membrane to affect properties of nAChRs18, and RIC-3 and NACHO, endoplasmic reticulum resident chaperones affecting functional expression of multiple nAChRs19,20.
Mutations in the C. elegans ric-3 gene were first identified in a screen for resistance to cholinesterase inhibitors21. Additional mutations were found by the Treinin lab in a screen for suppressors of neuronal degeneration caused by constitutive activity of a C. elegans nAChR, DEG-3/DES-222. Characterization of the C. elegans RIC-3 protein showed that it resides in the endoplasmic reticulum and is needed for maturation of multiple nAChRs. Further analysis showed conservation of ric-3’s sequence and function in evolution19. Importantly, RIC-3 enables functional expression of α7 nAChR when heterologously expressed in non-neuronal cells, even in cells that cannot normally express this receptor23. Additionally, brain expression patterns of mouse ric3 mRNA and CHRNA7 overlap, a result consistent with the suggestion that mammalian RIC-3 is required for α7 nAChR’s maturation in vivo19.
RIC-3 was shown to affect stability of unassembled nAChR subunits, assembly of subunits to form a nAChR, and trafficking of nAChRs24-29 (Figure 1 and below). In C. elegans loss of ric-3 gene function led to reduced plasma membrane expression of multiple nAChRs, demonstrating a positive, nAChR expression promoting function for RIC-322. In contrast, RIC-3’s effects on mammalian nAChRs are diverse. First, positive or negative effects on expression of functional receptors at the surface were demonstrated, depending on the identity of the nAChR and on the expression system used19,30. Recently we showed that these opposite effects on nAChR expression can be achieved using different RIC-3-to-receptor ratios, and that sensitivity of nAChR maturation to this ratio depends on the identity of the nAChR and on the specific RIC-3 isoform (different isoforms are produced by alternative splicing, see below)31. Second, RIC-3 was shown to affect different aspects of nAChR maturation depending on identity of the receptor’s subunits. Specifically, RIC-3 increased assembly and trafficking to the plasma membrane of α7 receptors, but not the quantity (likely to indicate stability) of α7 nAChR subunits; in contrast, RIC-3 increased quantity of α4 and β2 subunits while having no effect on their assembly into α4β2 receptors29. Thus, RIC-3 may interact with and affect different nAChRs differently.
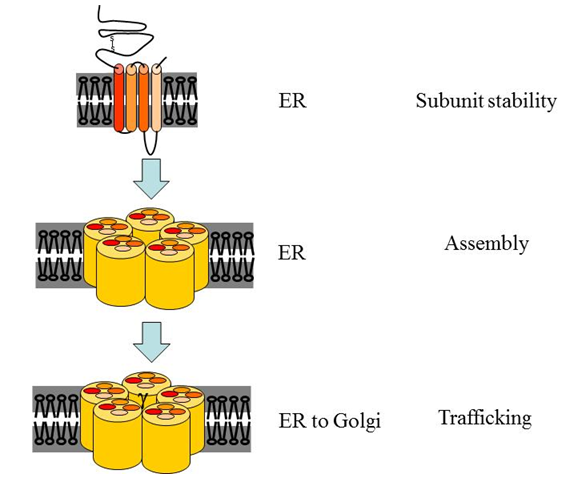
Figure 1: Steps in the maturation of nAChRs affected by RIC-3. Translation, subunit folding, and assembly of nAChRs occur in the endoplasmic reticulum (ER). Several aspects of this maturation process are affected by RIC-3. First, some unassembled subunits are stabilized by RIC-3. Second, RIC-3 increases assembly of some receptors and affects the subunit stoichiometry within the DEG-3/DES-2 receptor. Third, RIC-3 affects trafficking of fully assembled receptors from the ER to the Golgi on their way to the plasma membrane. Notably, effects on trafficking include both positive and negative effects.
Consistent with the suggestion that RIC-3 interacts differently with different nAChRs are results of structure-function analysis showing that mutations and deletions having no effect on one receptor strongly affect other receptors25,32. Moreover, RIC-3 was shown to affect properties (kinetics of desensitization and affinity to acetylcholine) of the C. elegans DEG-3/DES-2 nAChR. These effects were attributed to a RIC-3-dependent increase in the DEG-3 to DES-2 stoichiometry of this receptor, likely to be the result of preferential interaction between RIC-3 and the DEG-3 subunit, leading to its preferential insertion into the mature receptor24,25.
Mechanisms regulating RIC-3 expression and activity and their implications
As described above the nAChR family is large and diverse. Nevertheless, RIC-3 was shown to interact with and affect multiple nAChRs, differing in sequence and function19,30. But, these interactions and their outcome are variable as described above. This variability in function and interaction, combined with bioinformatics analysis, suggested that RIC-3 functions as an intrinsically disordered protein25 – proteins found to elicit opposing (inhibiting or activating) effects, even on the same partner molecule33. The variable and flexible interaction of intrinsically disordered proteins with their targets, and the extended protein interface dedicated to these interactions, favor regulation by alternative splicing and post-translational modifications, as these regulatory mechanisms can modify the interaction between the intrinsically disordered protein and its target thus regulating the outcome of the interaction34. Indeed, in C. elegans and mammals ric-3 expression, splicing, and activity are regulated, as described below and in Figure 2.
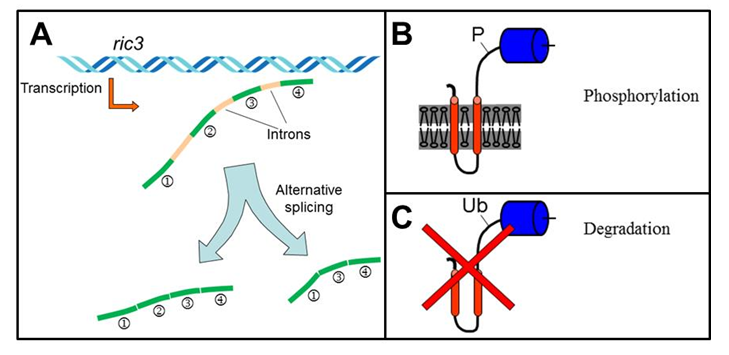
Figure 2: Mechanisms regulating RIC-3 expression and activity. A) In mammals ric-3 transcription and alternative splicing are regulated during inflammation with the gene's transcription being upregulated in Schizophrenia and Bipolar disorder patients. B) In C. elegans phosphorylation and dephosphorylation of RIC-3 affect its interactions with receptors. C) In C. elegans ubiquitination of RIC-3 is controlled in a cell specific manner by the CUL-3 adaptor, BATH-42, to protect cells for the deleterious effects of RIC-3 overexpression.
First, RIC-3 was shown to have either positive or negative effects on co-expressed receptors depending on the RIC-3-to-receptor ratio31. Thus mechanisms regulating RIC-3 expression or stability are likely to regulate cholinergic signaling via nAChRs. Indeed, our work identified mechanisms regulating RIC-3 quantity: In C. elegans BATH-42, an adaptor for CUL-3-dependent ubiquitin ligase, was shown to interact with RIC-3, an interaction leading to CUL-3-dependent degradation of RIC-335. And in murine macrophages we have shown that immune activation leads to a transient reduction in ric-3 mRNA31. Last, ric-3 expression was upregulated in postmortem brains of patients with Schizophrenia or Bipolar disorder36.
Second, phosphoproteomic analysis showed that in C. elegans RIC-3 is phosphorylated at multiple sites37. Analysis of one of these sites identified a Calcineurin-dependent signaling pathway regulating RIC-3 dephosphorylation and muscle excitability. Calcineurin is a calcium regulated phosphatase, thus we hypothesized that this pathway enables activity-dependent regulation of muscle excitability.
Third, vertebrates and invertebrates express multiple ric-3 transcripts, a result of alternative promoters, alternative splicing, and editing, reviewed by Treinin17. One conserved alternatively spliced RIC-3 isoform, lacking the conserved C-terminal coiled-coil domain, is defective for either the positive or negative effects of RIC-3 in a nAChR subtype dependent manner31.
RIC-3 and diseases of the Central Nervous System
RIC-3 was implicated in CNS diseases via expression studies and genetics. Expression analysis showed increased expression of ric3 in postmortem brains of patients with Schizophrenia and Bipolar disorder36. Genome-wide association studies demonstrated an association between single nucleotide polymorphisms upstream to the ric3 gene, which may affect its promoter, and MS or specific types of MS lesions38,39. And, RIC-3 was recently implicated in PD, as two different mutations affecting this protein were identified in patients. One mutation, P57T, was identified in a family with several members having late-onset PD with non-motor phenotypes; in this family all members carrying the RIC-3 mutation showed some disease phenotypes, with younger carriers showing weaker symptoms. The second mutation, V168L, was identified in an unrelated early-onset case40.
Multiple sclerosis (MS) is an inflammatory, demyelinating, immune-mediated disease of the CNS leading to axonal injury, axonal loss, and neurological deficits. Experimental Autoimmune Encephalomyelitis (EAE) serves as an animal model for the study of MS. The cholinergic anti-inflammatory pathway was suggested to influence MS progression. Indeed, cholinergic upregulation by acetylcholine esterase inhibitors or by nicotine reduced disease severity as well as causing immune-modulation, an effect mediated by the α7 nAChR41,42. Moreover, the cholinergic anti-inflammatory pathway was implicated in MS by studies comparing anti-inflammatory effects of nicotine on peripheral blood mononuclear cells (PBMCs) from patients vs. healthy donors43. This together with the known role of RIC-3 in α7 nAChR maturation suggests that RIC3 variants may affect MS via their effects on α7 nAChR maturation in immune cells. Importantly, we demonstrated that ric3 expression in immune cells is dynamically regulated following immune activation31. Moreover, recent unpublished results show that ric3 expression is transiently reduced in splenocytes and is gradually reduced in spinal cords during EAE progression (Yael Ben-David, unpublished). This suggests that regulation of RIC-3 expression is a means for regulating inflammation during EAE and MS.
PD is a neurodegenerative disease mainly affecting the motor system due to loss of dopaminergic neurons in the substantia nigra. Inflammation involving reactive astrocytes is known to contribute to progression of PD44. Involvement of nAChRs in PD was suggested by epidemiological studies showing reduced occurrence of this disease in smokers and altered expression of nAChRs in patients and disease models4,45. But, the mechanisms of action of nicotine and nAChRs in reducing PD are, as yet, unknown. Suggested mechanisms include neuroprotection46,47, effects on dopamine release48, and reduced neuro-inflammation resulting from activation of astrocytes or microglia49. Thus, the mechanisms of action of PD-associated RIC-3 mutations in this disease need to be identified. But, the demonstrated effects of these mutations on α7 nAChR surface expression40, the known role of this nAChR in regulating inflammation (above), and contribution of inflammation to neurodegenerative processes occurring in PD44,50, suggest a mechanism involving decreased activity of the cholinergic anti-inflammatory pathway as a mechanism explaining effects of both PD-associated RIC-3 mutations and nicotine on PD progression.
Conclusions
RIC-3 is an ER resident chaperone affecting maturation of multiple nAChRs including strong effects on functional expression of α7 nAChR. Several mechanisms regulate activity and expression of RIC-3, and the effects on nAChR maturation of these regulatory mechanisms depend on the nAChR subtype. RIC-3 has been implicated in progression of CNS diseases to which neuroinflammation and the cholinergic anti-inflammatory pathway are likely to contribute. Future work, therefore, should examine effects of genetic variants and regulatory mechanisms affecting RIC-3 expression and activity on the cholinergic anti-inflammatory pathway and neuroinflammation occurring in these diseases.
Conflict of interests
No conflict of interest to declare Funding: This research is funded by the United States-Israel Binational Science Foundation grant 2013055
References
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007; 47: 699-729.
- Kurzen H, Wessler I, Kirkpatrick CJ, et al. The Non-neuronal Cholinergic System of Human Skin. Horm Metab Res. 2007; 39: 2191-2194.
- Freedma, R, Hall M, Adler LE, et al. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995; 38: 22-33.
- Lange, KW, Wells FR, Jenner P, et al. Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson's disease. J Neurochem. 1993; 60: 197-203.
- Gotti C, Moretti M, Bohr I, et al. Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer's disease, Parkinson's disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol Dis. 2006; 23: 481-489.
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. Journal of pharmacology and experimental therapeutics. 1993; 26: 1455-1472.
- Coutrier S, Bertrand D, Matter JM, et al. A Neuronal nicotinic acetylcholine receptor subunit (α-7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron. 1990; 5: 847-856.
- Sequela P, Wadiche J, Dineley-Miller K, et al. W. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. The Journal of Neuroscience. 1993; 13: 596-604.
- Orr-Urtreger A, Göldner FM, Saeki M, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997; 17: 9165-9171.
- Deutsch SI, Burket JA, Benson AD, et al. The 15q13.3 Deletion Syndrome: Deficient α7-Containing Nicotinic Acetylcholine Receptor-Mediated Neurotransmission in the Pathogenesis of Neurodevelopmental Disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2016; 64:109-17.
- Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000; 405: 458-462.
- Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003; 421: 384-388.
- Yoshikawa H, Kurokawa M, Ozaki N, et al. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin Exp immunol. 2006; 146: 116-123.
- Park SY, Baik YH, Cho JH, et al. Inhibition of lipopolysaccharide-induced nitric oxide synthesis by nicotine through S6K1-p42/44 MAPK pathway and STAT3 (Ser 727) phosphorylation in Raw 264.7 cells. Cytokine. 2008; 44: 126-134.
- Sun Y, Li Q, Gui H, et al. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013; 23: 1270-1283.
- Treinin M. Role of the α7 nicotinic acetylcholine receptor and RIC-3 in the cholinergic anti-inflammatory pathway. Cent Nerv Sys Agents Med Chem. 201;. 17: 90-99.
- Treinin M. RIC-3 and nicotinic acetylcholine receptors: biogenesis, properties, and diversity. Biotechnology J. 2008; 3: 1539-1547.
- Miwa JM, Lester HA, Walz A. Optimizing cholinergic tone through lynx modulators of nicotinic receptors: implications for plasticity and nicotine addiction. Physiology. 2012; 27: 187-199.
- Halevi S, Yassin L, Eshel M, et al. Conservation within the RIC-3 gene family: effectors of nAChR expression. J Biol Chem. 2003; 278: 34411-34417.
- Gu S, Matta JA1, Lord B, et al. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron. 2016; 89: 948-955.
- Nguyen M, Alfonso A, Johnson CD, et al. Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics. 1995; 140L: 527-535.
- Halevi S, McKay J, Palfreyman M, et al. The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 2002; 21: 1012-1020.
- Williams ME, Burton B, Urrutia A, et al. Ric-3 promotes functional expression of the nicotinic acetylcholine receptor alpha7 subunit in mammalian cells. J Biol Chem. 2005; 280: 1257-1263.
- Cohen Ben-Ami H. Yassin L, Farah H, et al. RIC-3 affects properties and quantity of nicotinic acetylcholine receptors via a mechanism that does not require the coiled-coil domains. J Biol Chem. 2005; 280: 28053-28060.
- Cohen Ben-Ami H. Biala Y, Farah H, et al. Receptor and subunit specific interactions of RIC-3 with nicotinic acetylcholine receptors. Biochemistry. 2009; 48: 12329-12336.
- Alexander JK, Sagher D, Krivoshein AV, et al. Ric-3 promotes alpha7 nicotinic receptor assembly and trafficking through the ER subcompartment of dendrites. J Neurosci. 2010; 30: 10112-10126.
- Castillo M, Mulet J, Gutiérrez LM, et al. Dual role of the RIC-3 protein in trafficking of serotonin and nicotinic acetylcholine receptors. J Biol Chem. 2005; 280: 27062-27068.
- Cheng A, McDonald NA, Connolly CN. Cell surface expression of 5-hydroxytryptamine type 3 receptors is promoted by RIC-3. J Biol Chem. 2005; 280: 22502-22507.
- Dau A, Komal P, Truong M, et al. RIC-3 differentially modulates α4β2 and α7 nicotinic receptor assembly, expression, and nicotine-induced receptor upregulation. BMC Neurosci. 2013; 14: 47.
- Lansdell SJ, Gee VJ, Harkness PC, et al. RIC-3 enhances functional expression of multiple nicotinic acetylcholine receptor subtypes in mammalian cells. Mol Pharmacol. 2005; 68: 1431-1438.
- Ben-David Y, Mizrachi T, Kagan S, et al. RIC-3 expression and splicing regulate nAChR functional expression. Mol Brain. 2016; 9: 47.
- Biala Y, Liewald JF, Cohen Ben-Ami H, et al. The conserved RIC-3 coiled-Coil domain mediates receptor-specific Interactions with nicotinic acetylcholine receptors. Mol Biol Cell. 2009; 20: 1419-1427.
- Tompa P, sz CS, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005; 30: 484-489.
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015; 16: 18-29.
- Shteingauz A, Cohen E, Biala, Y, et al. The BTB-MATH protein BATH-42 interacts with RIC-3 to regulate maturation of nicotinic acetylcholine receptors. J Cell Sci. 2009; 122: 807-812.
- Severance EG Yolken RH. Lack of RIC-3 congruence with beta2 subunit-containing nicotinic acetylcholine receptors in bipolar disorder. Neurosci. 2007; 148: 454-460.
- Zielinska DF, Gnad F, Jedrusik-Bode M, et al. Caenorhabditis elegans has a phosphoproteome atypical for metazoans that is enriched in developmental and sex determination proteins. J Proteome Res. 2009; 8: 4039-4049.
- Gourraud PA, Sdika M, Khankhanian P, et al. A genome-wide association study of brain lesion distribution in multiple sclerosis. Brain. 2013; 136: 1012-1024.
- Wang JH, Pappas D, De Jager PL, et al. Modeling the cumulative genetic risk for multiple sclerosis from genome-wide association data. Genome Med. 2011; 3: 3.
- Sudhaman S. Muthane UB, Behari M, et al. Evidence of mutations in RIC3 acetylcholine receptor chaperone as a novel cause of autosomal-dominant Parkinson's disease with non-motor phenotypes. J Med Genet. 2016; 53: 559-566.
- Nizri E, Irony-Tur-Sinai M, Lory O, et al. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol. 2009; 183: 6681-6688.
- Nizri E, Hamra-Amitay Y, Sicsic C, et al. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacol. 2006; 50: 540-547.
- Reale M, Di Bari M2, Di Nicola M, et al. Nicotinic receptor activation negatively modulates pro-inflammatory cytokine production in multiple sclerosis patients. Int. Immunopharmacol. 2015; 29: 152-157.
- Booth H DE, Hirst WD, Wade-Martins R. The Role of Astrocyte Dysfunction in Parkinson's Disease Pathogenesis. Trends Neurosci. 2017; 40: 358-370.
- Li, X., Li, W., Liu, G., et al. Association between cigarette smoking and Parkinson's disease: A meta-analysis. Arch Gerontol Geriatr. 2015; 61: 510-516.
- Stevens TR, Krueger SR, Fitzsimonds RM, et al. Neuroprotection by nicotine in mouse primary cortical cultures involves activation of calcineurin and L-type calcium channel inactivation. J Neurosci. 2003; 23: 10093-10099.
- Srinivasan R, Henley BM1, Henderson BJ, et al. Smoking-Relevant Nicotine Concentration Attenuates the Unfolded Protein Response in Dopaminergic Neurons. J Neurosci. 2016; 36: 65-79.
- Quik M, Perez XA Grady SR. Role of α6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem Pharmacol. 2011; 82: 873-882.
- Liu Y, Hu J, Wu J, et al. α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation 2012; 9: 98.
- Liddelow SA, Guttenplan KA1, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017; 541: 481-487.