
SARS-CoV-2 Subversion of the Antiviral Interferon Alpha-Response of Lung Macrophages?
Malgorzata Kloc*1,2,3, Rafik M. Ghobrial1,2, Jacek Z Kubiak*4,5
1The Houston Methodist Research Institute, Houston, Texas 77030, USA
2The Houston Methodist Hospital, Department of Surgery, Houston, Texas, USA
3The University of Texas, M.D. Anderson Cancer Center, Department of Genetics, Houston Texas, USA
4Laboratory of Regenerative Medicine and Cell Biology, Military Institute of Hygiene and Epidemiology (WIHE), Warsaw, Poland
5UnivRennes, UMR 6290, CNRS, Institute of Genetics and Development of Rennes, Cell Cycle Group, Faculty of Medicine, Rennes, France
Abstract
The interferons (IFNs) are the main antiviral immune factors. Currently, various IFNs therapies are used for the treatment of human immunodeficiency virus (HIV), hepatitis B (HBV), and hepatitis C (HCV), cancer, and autoimmune diseases. Recently, it has been suggested that IFN-α therapy should be used to lessen the respiratory symptoms in the SARS-CoV-2 virus- infected (COVID-19) patients. The SARS-CoV-2 enters the cells by binding to the Angiotensin-converting enzyme 2 (ACE2), which by recognizing the spike S1 protein of the virus, acts as a virus receptor. Because the expression of ACE2 is induced by IFN-α, the SARS-CoV-2 virus may exploit the anti-viral response by subverting the IFN functions to further its own propagation and infectability. We discuss here how the SARS-CoV-2 may also subvert the immune response of the lung macrophages, which also express ACE2, to exacerbate the severity of the COVID-19 respiratory symptoms.
The interferons (IFNs) are the principal signaling molecules (cytokines) released by the immune system to fight viral infections. Because of their potency and a wide spectrum of cellular targets, they also result in the flu-like side effects, such as chills, fever, headache, and muscle pain. The IFNs are the soluble glycoproteins categorized into type I (containing 12 subtypes of IFN-α, IFN-β IFN-ε, IFN-κ, and IFN-ω), type II (containing only IFN-γ), and type III (IFN-λ 1, IFN-λ 2, and IFN-λ 3)1. All these interferons activate the anti-viral response in all cells, which express the respective receptors. The type I IFNs bind to the IFNα/β receptor (IFNAR) that is present in the majority of cells, including macrophages, and through the tyrosine kinase 2 (TYK2), Janus kinase 1 (JAK1), and the signal transducers and activators of transcription (STATs) induce the formation of the interferon-stimulated gene factor 3 (ISGF3) complex. When this complex translocates to the cell nucleus, it binds the interferon-stimulated response elements (ISREs) in the promoter region of interferon responsive genes (ISGs), and switches on the transcription of the antiviral genes1. During microbial infection, majority of cells produce type I interferons (IFNs). Macrophages and dendritic cells (DCs), produce type I IFNs after sensing a pathogen. Alveolar macrophages produce copious quantities of IFNβ and less IFNα. The dendritic cells (DC) and especially plasmacytoid DCs (pDCs) synthesize mainly IFNα, while the non-immune cells, such epithelial cells, and fibroblasts synthesize mainly IFNβ. Type II (IFN-γ) is produced by many types of immune cells including T cells, NK cells, and macrophages2. The type II (IFN-γ) receptors are expressed mainly in the T cells, natural killer cells, and natural killer T (NKT) cells that are the T cells with killer cells activities. The binding of the IFN-γ to its receptor (IFN-γR1 and IFN-γR2 chains) activates JAK1, JAK2, and STAT signaling pathways. When the, phosphorylated by JAK1, STAT1 homodimerizes and translocates to the nucleus, it binds to promoters that contain an interferon-γ-activated site (GAS), and activates transcription of the relevant immune response genes. Another, more direct, route of transcriptional activation involves the binding of the ligand-binding subunit (alpha) of the IFN-γ receptor (IFNGR1) to the phosphorylated STAT1, translocation of the complex to the cell nucleus, binding to the GAS promoter region of the responsive genes, and activation of their transcription1. The type III interferons (IFN-λs) are expressed by many different cell types, but the expression of IFNRλ receptors are mainly limited to the epithelial, and epithelial-derived cells. There are older studies showing that mesothelial peritoneal cells respond to the IFNRλ that suggests that also the pleural cavities cells express IFNRλ receptors3,4. The immune cells such as macrophages, dendritic cells, B cells, and T cells, express a short variant of the IFNRλ receptor, which is only weakly activated by the IFN-λs. The binding of INFλ to its receptor recruits IL-10R2, and the resulting complex activates, (similar to IFNs type I) JAK/STAT signaling and regulates transcription of the target genes1. The IFNs do not directly kill the virus but their anti-viral activity relies on the activation of the transcription of genes whose protein products directly inhibit replication of the virus in the infected cells, and also on its immunomodulatory effect on the various subsets of the immune cells, which kill and remove infected cells, and, thus, restrict the infection. Currently, various IFNs therapies are used for the treatment of human immunodeficiency virus (HIV), hepatitis B (HBV), and hepatitis C (HCV), cancer, and autoimmune diseases. Recently performed immunoprofiling of COVID-19 patients showed that 1 out of 5 critically ill patients had a low or undetectable level of IFN-α, which led to the conclusion that the COVID-19 patients should be screened for the level of IFN I, and those with defective IFN-α response should undergo the IFN-α therapy5.
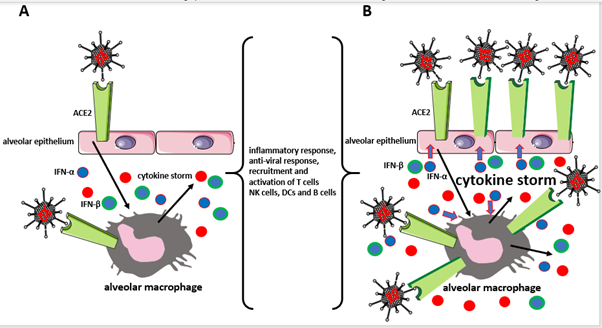
Figure 1: Two scenarios of IFN-α function in the lungs infected with the SARS-CoV-2 virus.
A) IFN-α is only a part of the inflammatory/anti-viral response, and B) IFN-α is highjacked by the virus to produce more ACE2 that serves as the virial receptors, increasing virus infectability.
In scenario A, the SARS-CoV-2 infects epithelial cells of lung alveoli using ACE2 as its receptor. The inflammatory signals from the infected epithelial cells activate immune/ anti-viral responses in the alveolar macrophages. The cytokines, including IFN-α and IFN-β produced by macrophages and epithelial cells also recruit/activate immune/antiviral response in other types of immune cells (T cells, NK, DCs, and B cells). Because besides the epithelial cells the alveolar macrophages also express the ACE2, they are also infected by the virus exacerbating the immune response and causing damaging cytokine storm. Eventually, the avalanche of the produced inflammatory/anti-viral factors leads to the acute respiratory distress syndrome (ARDS) in the COVID-19 patient.
In scenario B, the IFN-α and IFN-β produced in response to SARS-CoV-2 infection, not only fights the virus but also induces the transcription of the ACE2 gene in the epithelial cells and macrophages, leading to the formation of more ACE2 receptors. This increases the infectability of the virus, but also further exacerbates the damaging already cytokine storm and further increases the severity of ARDS in COVID-19 patients’ lungs.
However, this approach may be problematic, because another, also published in 2020 study, suggests that the SARS-CoV-2 virus that causes the current world-wide COVID-19 pandemic may exploit the anti-viral response by subverting the IFN functions to further own propagation and infectability6. We postulate here that SARS-CoV-2 may also subvert the immune response of the lung macrophages exacerbating the severity of the COVID-19 respiratory symptoms.
The SARS-CoV-2 is the RNA virus that, similar to SARS-COV and NL63, enters the cells by binding to the Angiotensin-converting enzyme 2 (ACE2), which by recognizing the spike S1 protein of the virus, acts as a virus receptor7,8. Besides serving as the virus receptor, the main, enzymatic, function of ACE2, which is the zinc metallopeptidase angiotensin-converting enzyme 2, is the catalytic conversion of the vasopressor angiotensin II to the vasodilator angiotensin-(1–7) and, thus, regulation of the blood pressure and heart functions.
One of the major and most dangerous symptoms of COVID-19 is acute respiratory distress syndrome (ARDS), which causes death in approximately 15% of patients. The main portals for the entry of SARS-CoV-2 to the body using the ACE2 receptors are the nasal epithelial cells. However, the single-cell RNA expression profiling studies showed recently that the ACE2 is not only abundant in the airway epithelial cells, the epithelial cells of lung alveoli, but also in the lung macrophages9. There are two major types of lung macrophages, the alveolar and parenchymal. After receiving the distress (cytokine) signals from the virally infected lung epithelial cells, these macrophages try to eliminate the virus by mounting a rapid immune response: destroying (phagocyting) the infected cells and releasing the massive amount of immune defense/signaling factors such as various cytokines and IFN-α, and IFN-γ2,10, which activate and recruit other immune cells to join in the fight against the virus. This is usually effective in eliminating the infection and resolving lung inflammation in the fight against common respiratory viruses. However, in SARS-CoV-2 infection, because the lung macrophages express ACE2 receptors, they also become infected11, which exacerbates their immune response causing the release of abnormally high amounts of cytokines and other inflammatory molecules. This, in turn, causes the so-called cytokine storm resulting in a lethal ARDS. This, by itself, would be worrisome enough, but there is also another, very troubling finding. The single-cell RNA-seq datasets analysis showed recently that the expression of the ACE2 gene in human (but not in mouse) airway epithelial cells is stimulated by the IFN-α6. Thus, the massive production of IFN-α during the anti-viral immune response in the infected lungs would increase the number of ACE2 receptors for the SARS-CoV-2 virus and increase the rate of infection. Because the alveolar macrophages are the main producers of IFN-α in lung infections with many RNA viruses10 we postulate that the released IFN-α not only upregulates ACE2 receptors for the SARS-CoV-2 in the respiratory epithelial cells but also in the lung macrophages that, in turn, increases macrophage infection with the virus, and even more exacerbates the macrophage-derived cytokine storm and severity of ARDS. Currently there are nearly 4000 COVID-19 clinical trials (listed on the website: https://clinicaltrials.gov/ct2/results?cond=COVID-19) there is one clinical trial ( http://www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=43775) with pegylated IFN alpha-2b. Limited studies indicate that the intranasal delivery of IFN-β is effective in reducing the viral load and providing some protection against COVID-19 infection12. Although the alveolar macrophages produce more IFN-β than IFN-α, which have both pro- and anti-inflammatory activities, these two responses are deregulated in the lungs of COVID-19 patients. A cytokine storm may reflect a sum of these two responses that may vary among the individual patients. Although some limited or uncontrolled studies indicate the benefits of IFN-α treatment of COVID-19 patients13, further studies on the correlation between IFN-α, ACE2 upregulation, lung macrophages, and SARS-CoV-2 infection, and also caution in the therapeutic use of IFN-α as the anti-SARS-CoV-2 medication are necessary.
Acknowledgements
We acknowledge the support from the Polish Ministry of National Defense project “KoÅciuszko" # 5508/2017/DA to JZK. We also acknowledge that some of the images used to make figure were from the Servier Medical ART: SMART, smart.servier.com.
References
- Lin F-C, and Young HA (2014). Interferons: Success in Anti-Viral Immunotherapy. Cytokine Growth Factor Rev. 25:369-76. doi: 10.1016/j.cytogfr.2014.07.015.
- Darwich L, Coma G, Peña R, Bellido R, Blanco EJ, Este JA, Borras FE, Clotet B, Ruiz L, Rosell A, Andreo F, Parkhouse RM, Bofill M. (2009). Secretion of interferon-gamma by human macrophages demonstrated at the single-cell level after costimulation with interleukin (IL)-12 plus IL-18. Immunology. 126:386-93. doi: 10.1111/j.1365-2567.2008.02905.x.
- Topley, N., R. K. Mackenzie, and J. D. Williams. 1996. Macrophages and mesothelial cells in bacterial peritonitis. Immunobiology 195:563-573.
- Li, F. K., et al. 1998. Leukocyte migration across human peritoneal mesothelial cells is dependent on directed chemokine secretion and ICAM-1 expression. Kidney Int. 54:2170-2183
- Trouillet-Assant S, Viel S, Gaymard A, Pons S, Richard J-C, Perret M, Villard M, Brengel-Pesce K, Lina B, Mezidi M, Bitker L, Belot A. (2020). Type I IFN immunoprofiling in COVID-19 patients. Journal of Allergy and Clinical Immunology, 29 April 2020, doi: 10.1016/j.jaci.2020.04.029
- Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN. Et al (2020). SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 181: 1016–1035.e19. doi: 10.1016/j.cell.2020.04.035
- Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. (2020). Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448.
- Turner AJ, Hiscox JA, Hooper NM. (2004). ACE2: from vasopeptidase to SARS virus receptor. TRENDS in Pharmacological Sciences 25: 291–294.
- Zhao Y, Zhao Z, WangY, ZhouY, Ma Y, Zuo W. (2020). Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. bioRxiv preprint doi: https://doi.org/10.1101/2020.01.26.919985. Posted January 26, 2020.
- Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E,Aozasa K, Kawai T, Akira S. (2007). Alveolar Macrophages Are the Primary Interferon-a Producer in Pulmonary Infection with RNA Viruses. Immunity 27, 240–252.
- Abassi Z, Knaney Y, Karram T, 3 Heyman SN. (2020). The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 05 June 2020. https://doi.org/10.3389/fimmu.2020.01312
- Lee, J.S., Shin, E. The type I interferon response in COVID-19: implications for treatment. Nat Rev Immunol (2020).
- Zhou Q, Chen V, Shannon CP, Wei X-S, Xiang X et al. Interferon-α2b Treatment for COVID-19 Front Immunol. 2020; 11: 1061. Published online 2020 May 15. doi: 10.3389/fimmu.2020.01061