
The Neuro-Psychological Axis of Smoking-Associated Cancer
Hildegard M Schuller*
Department of Biomedical & Diagnostic Sciences, College of Veterinary Medicine, University of Tennessee, 2407 River Drive, Knoxville, TN 37996, USA
Abstract
This mini-review summarizes current knowledge on similarities and synergism between smoking and psychological stress-induced modulations of growth stimulating and inhibiting regulatory networks in epithelial cells and epithelial cancers with emphasis on cancer stimulating neurotransmitters and their receptors as well as cancer inhibiting γ-aminobutyric acid (GABA) and opioids. Hyperactive cAMP signaling downstream of beta-adrenergic receptors (β-ARs) has been identified as the driving force of most smoking-associated cancers by numerous preclinical studies and psychological stress intensifies these effects while experimental stress reduction inhibits. The integration of cAMP reduction via stress reduction by pharmacological and psychological means such as psychotherapy, relaxation meditation and yoga into any cancer treatment strategy is recommended.
Introduction
Smoking is a documented risk factor for numerous human cancers, including cancer of the lungs, larynx, esophagus, stomach, breast, pancreas, colon, prostate and bladder1-3, with a particularly strong etiological association between smoking and cancer of the larynx , lungs4 and pancreas5. Research into the mechanisms of tobacco-associated carcinogenesis has identified several powerful carcinogens in tobacco smoke, including polycyclic aromatic hydrocarbons (predominantly benzo[a]pyrene) and the nicotine derived nitrosamines N’-nitrosonornicotine (NNN) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (N’nitrosonicotine ketone, NNK)1.
Research into the mechanisms of action of tobacco carcinogens has identified interactions of their metabolites with DNA that result in the formation of inactivating mutations in the tumor suppressor gene p536 and in mutations in the k-ras gene6 that sensitize the gene to its upstream stimulators7. Both mutations are frequently expressed in tobacco-associated human cancers8-9. Moreover, NNN and NNK are agonists for nicotinic acetylcholine receptors (nAChRs) with significantly higher affinity than their physiological agonist acetylcholine or nicotine10 and NNK is additionally an agonist for beta-adrenergic receptors (β-ARs) with significantly higher affinity than their physiological agonists epinephrine (Epi) and norepinephrine (Nor)11. In light of the ubiquitous expression of nAChRs and β-ARs in mammalian cells12-13, these findings prompted research into the potential role of neurotransmitter receptors of the nicotinic cholinergic and beta-adrenergic families in the development, progression and resistance to therapy of cancer.
Regulation of cancer by the nAChR-mediated release of neurotransmitters
The excitatory neurotransmitters acetylcholine, serotonin, glutamate, dopamine, Epi and Nor are not only synthesized and released by the brain and the autonomic nervous system but also by normal epithelial cells and epithelial cancers12, 14-17 and Nor and Epi in addition by the adrenal gland18. Acetylcholine is the physiological agonist for nAChRs and opens their ion channel upon binding to the receptor, resulting in membrane depolarization that triggers the opening of voltage-gated Ca2+-channels. In turn, this allows for the influx of Ca2+ ions, causing the release of neurotransmitters19.
Early in vitro studies have shown that binding of nicotine to the α7nAChR regulates the autocrine regulation of cell proliferation by serotonin in small cell lung cancer cells20. In addition, it has been shown that increases in systemic serotonin stimulated the growth of colon cancer allografts in mice by inducing angiogenesis21.
More recent investigations have shown that the α7nAChR regulates the release of Epi and Nor in vitro from cells of normal small airway epithelium, lung adenocarcinoma16, pancreatic duct epithelia and pancreatic ductal adenocarcinoma15, gastric cancer22, colon cancer23, and urothelial bladder cancer24 and induces their proliferation and migration via this autocrine mechanism. Moreover, all of these cancers as well as prostate cancer, ovarian cancer, breast cancer, and hemangiosarcoma are stimulated in their growth by exposure to exogenous Epi, Nor or synthetic beta-adrenergic agonist while the non-selective beta-blocker propranolol inhibits the autocrine and exogenous stimulation of these cancers24-26. In addition, it has been shown in adenocarcinomas of the lung and pancreas that NNK has identical cancer-stimulating effects as Epi and Nor by binding as an agonist to β-ARs and that propranolol inhibited these responses11, 25.
The amino acid neurotransmitter glutamate is synthesized and released by numerous cancers and stimulates their proliferation and migration, including cancer of the pancreas, prostate, breast and adenocarcinoma of the lungs26. In turn, the release of glutamate is regulated by the α7nAChR27. Moreover, the α7nAChR regulates the release of the catecholamine neurotransmitter dopamine and its receptors that are expressed in many cancers and can have cancer stimulating as well as inhibitory effects pending on the expression levels of receptors of the D1-like family which increase cAMP signaling via the G-protein Gsq or receptors of the D2-like family that are coupled to the inhibitory G-protein Gi and inhibit cAMP formation28-29.
Antagonistic effects of receptors coupled to the stimulatory G protein Gs and receptors coupled to the inhibitory G protein Gi
Beta-adrenergic receptors are coupled to the stimulatory G protein Gs. Activation of Gs by binding of an agonist to the receptor activates the enzyme adenylyl cyclase that catalyzes the formation of intracellular cyclic adenosine monophosphate (cAMP) which in turn activates protein kinase A (PKA)30. Increased intracellular cAMP and activated PKA stimulate the release of epidermal growth factor (EGF)25, 31, vascular endothelial growth factor (VEGF)32-33 and arachidonic acid (AA)11 from the cancer cells and from fibroblasts, macrophages and endothelial cells in the stroma that constitutes the cancer micro-environment34-35. Each one of these released products stimulates the growth, metastatic potential and resistance to therapy of cancer.
Receptors coupled to the inhibitory G protein Gi and their endogenous agonists are the physiological inhibitors of Gs-coupled receptors. In accord with this function, it has been shown that the inhibitory neurotransmitter γ-aminobutyric acid (GABA) has tumor suppressor function via Gi-coupled GABAB receptors in vitro and in animal models for adenocarcinoma of the lung36 and pancreas37. Cancer stem cells isolated from pancreatic cancer cell lines stimulated their self-renewal by the autocrine release of Epi and Nor that activated beta-adrenergic signaling and these effects were blocked by treatment with GABA38. In addition, the opioid dynorphin B inhibited the self-renewal of cancer stem cells from lung adenocarcinomas via their Gi-coupled opioid receptors39. Similarly, the synthetic opioid methadone has strong inhibiting effects on numerous cancers40-41. Moreover, preclinical studies have shown that the endogenous cannabinoid system is activated by binding of exogenous cannabinoids (medical marihuana, synthetic cannabinoids) to Gi-coupled cannabinoid receptors, resulting in growth inhibition and improved response to therapy of lung adenocarcinoma, colon cancer and glioblastoma42-44.
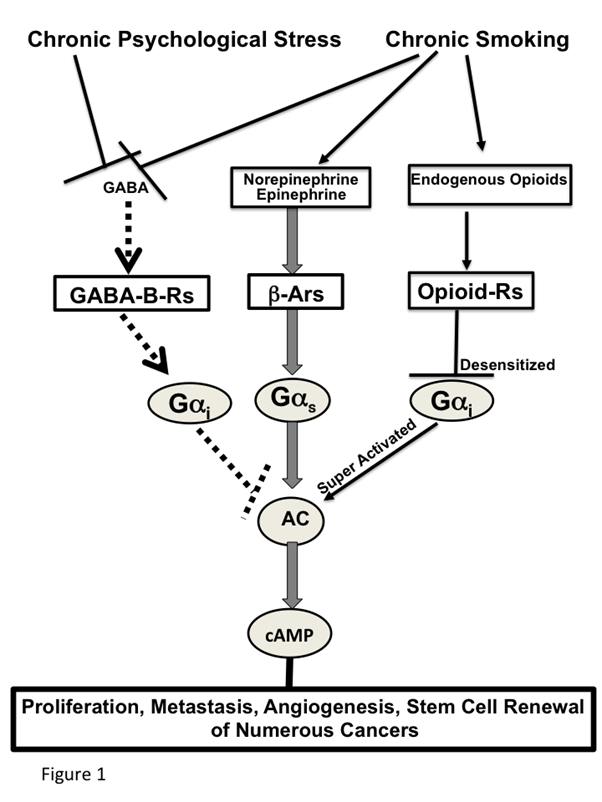
Effects of smoking and chronic psychological stress on cancer stimulating and inhibiting networks (Figure 1)
Smoking and chronic psychological stress each induce the nAChR-regulated release of Epi and Nor, thereby increasing their systemic levels45. In turn, this creates an environment that supports the development and progression of numerous cancers for which Epi and Nor act as strong growth factors. Smoking and chronic psychological stress additionally suppress the GABA system46-47, thus depriving the body of the physiological inhibitor of Epi and Nor-induced cancer stimulation. Acute exposure to nicotine stimulates the nAChR-mediated release of endogenous opioids above physiological levels48. Similar to opioid addiction, the continued exposure to unphysiologically high opioid levels during chronic nicotine-induced nicotine addiction and withdrawal desensitizes the Gi-coupled opioid receptors49, resulting in a reactive super activation of adenylyl cyclase/cAMP signaling48, 50. On the other hand, stress reduction51 and positive emotions52 decrease the levels of stress neurotransmitters while simultaneously increasing the levels of GABA and endogenous opioids within their phsyiologal range, thereby restoring cAMP homeostasis. The strong influence of these neuropsychological factors on cancer development and progression has been documented by preclinical investigations which have reported significant cancer-stimulating effects of experimentally induced stress on cancer of the lungs53, pancreas37, breast54-55 and ovary56 whereas stress reduction by species appropriate environmental enrichment significantly reduced the development and progression of mouse xenografts from lung adenocarcinomas39.
Conclusions and future directions
The addictive properties of smoking have been extensively investigated. The focus of that research has been on nicotine-induced changes in nAChR-mediated brain neurotransmission characterized by hyperactivity of excitatory neurotransmitters accompanied by suppression of their physiological inhibitors, the GABA and endogenous opioid systems, and the resulting psychological responses associated with addiction and withdrawal symptoms. However, the fact that smoking also causes cardiovascular disease by elevating systemic Epi and Nor levels due to their increased release from the adrenal gland and sympathetic nervous system strongly suggests that smoking-induced modulations in nAChR expression and function are not restricted to the brain, where they cause addiction, but instead occur universally in non-neuronal cells and tissues as well where their altered functions cause non-neuronal diseases. As is summarized in this mini-review, epithelial cells express nAChR-regulated autocrine signal transduction pathways that maintain the balance between excitatory neurotransmitters that stimulate cell proliferation and GABA which inhibits. The same changes that cause nicotine addiction when occurring in brain nAChRs cause systemic and epithelial hyperactivity of cancer stimulating neurotransmitters while suppressing inhibitory GABA. The release of Epi and Nor from cancer cells and the sympathetic nervous system is predominantly stimulated by the homomeric α7nAChR14 which does not undergo long-lasting desensitization in response to chronic nicotine exposure57. By contrast, the heteromeric α4β2nAChRs that regulates GABA release from epithelial cells and epithelial cancers desensitizes in response to chronic nicotine, resulting in suppressed GABA release58. The unrestricted growth of cancer cells is further supported by the systemic increase in Nor and Epi and simultaneous suppression of the endogenous opioid system and GABA system caused by smoking and chronic psychological stress. The resulting beta-adrenergic receptor hyperactivity additionally impairs the immune system,via cyclooxynenase-2-mediated suppression of CD8+ T cell responses59, an effect caused by the beta-adrenergic stimulation of arachidonic acid release in cancer cells11.
Current therapeutic strategies of cancer therapy aim to destroy existing cancer cells by chemotherapy, radiation and immunotherapy. These treatments shrink existing tumors, thereby often rendering them surgically resectable, resulting in significant increases in overall survival times. However, they do not remove the imbalance in cancer stimulating and inhibiting regulatory networks characterized by hyperactive cAMP signaling that is caused by smoking and chronic psychological stress, which often work synergistically. Accordingly, the majority of cancers eventually relapse. A major goal of adjuvant cancer therapy aimed at preventing the formation of new cancer cells via self-renewing stem cells and the associated progression, resistance to therapy and cancer relapse should therefore be the restoration of cAMP homeostasis. General beta-blockers such as propranolol used successfully for the long-term management of cardiovascular disease, nutritional GABA supplements or over the counter valerian extracts that stimulates the endogenous synthesis of GABA (both widely used as sleep aids and anxyolytics), positive allosteric modulators of the GABAB-R used for the management of drug addiction as well as opioids used for anesthesia, analgesia, cough suppression and the management of drug addiction should all be explored in clinical trials as adjuvant therapy of cancer. Finally, the powerful cancer stimulating effects of psychological stress and cancer inhibiting effects of psychological well -being cannot be over-emphasized. Stress reduction/relaxation by psychotherapy, relaxation meditation and yoga should be an essential component of any cancer treatment plan.
Funding Information
Funding for the original research publications by the author referenced in this review has been acknowledged in the original publications.
References
- Leon ME, Peruga A, McNeill A, et al. European Code against Cancer, 4th Edition: Tobacco and cancer. Cancer Epidemiol. 2015; 39 Suppl 1: S20-33.
- Catsburg C, Miller AB, Rohan TE. Active cigarette smoking and risk of breast cancer. Int J Cancer. 2015; 136 (9): 2204-9.
- De Nunzio C, Andriole GL, Thompson IM Jr, et al. Smoking and prostate cancer: A systematic review. Eur Urol Focus. 2015; 1 (1): 28-38.
- Alberg AJ, Shopland DR, Cummings KM. The 2014 Surgeon General's report: commemorating the 50th Anniversary of the 1964 Report of the Advisory Committee to the US Surgeon General and updating the evidence on the health consequences of cigarette smoking. Am J Epidemiol. 2014; 179 (4): 403-12.
- Maisonneuve P, Lowenfels AB. Risk factors for pancreatic cancer: a summary review of meta-analytical studies. Int J Epidemiol. 2015; 44 (1): 186-98.
- Hecht SS. Progress and challenges in selected areas of tobacco carcinogenesis. Chem Res Toxicol. 2008; 21 (1): 160-71.
- Logsdon CD, Lu W. The Significance of Ras Activity in Pancreatic Cancer Initiation. Int J Biol Sci. 2016; 12 (3): 338-46.
- Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002' 21 (48): 7435-51.
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72 (10): 2457-67.
- Schuller HM, Orloff M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol. 1998; 55 (9): 1377-84.
- Schuller HM, Tithof PK, Williams M, et al. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999; 59 (18): 4510-5.
- Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008; 154 (8): 1558-71.
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002; 3 (9): 639-50.
- Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009; 9 (3): 195-205.
- Al-Wadei MH, Al-Wadei HA, Schuller HM. Pancreatic Cancer Cells and Normal Pancreatic Duct Epithelial Cells Express an Autocrine Catecholamine Loop that Is Activated by Nicotinic Acetylcholine Receptors alpha3, alpha5, and alpha7. Mol Cancer Res. 2012; 10 (2): 239-49.
- Al-Wadei HA, Al-Wadei MH, Masi T, et al. Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells. Lung Cancer. 2010: 69 (1): 33-9.
- Tang J, Li Z, Lu L, et al. beta-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol. 2013; 23 (6 Pt B): 533-42.
- Lymperopoulos A, Brill A, McCrink KA. GPCRs of adrenal chromaffin cells & catecholamines: The plot thickens. Int J Biochem Cell Biol. 2016; 77 (Pt B): 213-9.
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006; 27 (9): 482-91.
- Cattaneo MG, Codignola A, Vicentini LM, et al. Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res. 1993; 53 (22): 5566-8.
- Nocito A, Dahm F, Jochum W, et al. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008; 68 (13): 5152-8.
- Shin VY, Wu WK, Chu KM, et al. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol Sci. 2007; 96 (1): 21-9.
- Wong HP, Yu L, Lam EK, et al. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharmacol. 2007; 221 (3): 261-7.
- Papu John AM, Banerjee J, Schuller HM. In Regulation of urothelial bladder cancer by nicotine and stresss neurotrasnmitters, 105th AACR Annual Meeting, San Diego, CA, American Association for Cancer Research: San Diego, CA, 2014.
- Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol. 2005; 131 (10): 639-48.
- Stepulak A, Rola R, Polberg K, et al. Glutamate and its receptors in cancer. J Neural Transm (Vienna). 2014; 121 (8): 933-44.
- Ryu IS, Kim J, Seo SY, et al. Behavioral changes after nicotine challenge are associated with alpha7 nicotinic acetylcholine receptor-stimulated glutamate release in the rat dorsal striatum. Sci Rep. 2017; 7 (1): 15009.
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004; 24 (3): 165-205.
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002; 296 (5573): 1636-9.
- Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf). 2007; 190 (1): 9-19.
- Grau, M, Soley M, Ramirez I. Interaction between adrenaline and epidermal growth factor in the control of liver glycogenolysis in mouse. Endocrinology. 1997; 138 (6): 2601-9.
- Al-Wadei HA, Al-Wadei MH, Ullah MF, et al. Celecoxib and GABA cooperatively prevent the progression of pancreatic cancer in vitro and in xenograft models of stress-free and stress-exposed mice. PLoS One. 2012; 7 (8): e43376.
- Madden KS, Szpunar MJ, Brown EB. beta-Adrenergic receptors (beta-AR) regulate VEGF and IL-6 production by divergent pathways in high beta-AR-expressing breast cancer cell lines. Breast Cancer Res Treat. 2011; 130 (3): 747-58.
- Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015; 368 (1): 7-13.
- Mantovani A, Marchesi F, Malesci A, et al. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017; 14 (7): 399-416.
- Schuller HM, Al-Wadei HA, Majidi M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis. 2008; 29 (10): 1979-85.
- Schuller HM, Al-Wadei HA, Ullah MF, et al. Regulation of pancreatic cancer by neuropsychological stress responses: a novel target for intervention. Carcinogenesis. 2012; 33 (1): 191-6.
- Al-Wadei MH, Banerjee J, Al-Wadei HA, et al Nicotine induces self-renewal of pancreatic cancer stem cells via neurotransmitter-driven activation of sonic hedgehog signalling. Eur J Cancer. 2016; 52: 188-96.
- Banerjee J, Papu John AM, Schuller HM. Regulation of nonsmall-cell lung cancer stem cell like cells by neurotransmitters and opioid peptides. Int J Cancer. 2015; 137 (12): 2815-24.
- Friesen C, Hormann I, Roscher M, et al. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle. 2014; 13 (10): 1560-70.
- Michalska M, Katzenwadel A, Wolf P. Methadone as a "Tumor Theralgesic" against Cancer. Front Pharmacol. 2017; 8: 733.
- Ramer R, Bublitz K, Freimuth N, et al. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 2012; 26 (4): 1535-48.
- Chakravarti B, Ravi J, Ganju RK. Cannabinoids as therapeutic agents in cancer: current status and future implications. Oncotarget. 2014; 5 (15): 5852-72.
- Romano B, Borrelli F, Pagano E, et al Inhibition of colon carcinogenesis by a standardized Cannabis sativa extract with high content of cannabidiol. Phytomedicine. 2014; 21 (5): 631-9.
- Robinson JD, Cinciripini PM. The effects of stress and smoking on catecholaminergic and cardiovascular response. Behav Med. 2006; 32 (1): 13-8.
- D'Souza MS. Neuroscience of nicotine for addiction medicine: novel targets for smoking cessation medications. Prog Brain Res. 2016; 223: 191-214.
- Hu W, Zhang M, Czéh B, et al. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology. 2010; 35 (8): 1693-707.
- Kishioka S, Kiguchi N, Kobayashi Y, et al. Nicotine effects and the endogenous opioid system. J Pharmacol Sci. 2014; 125 (2): 117-24.
- McCarthy MJ, Zhang H, Neff NH, et al Desensitization of delta-opioid receptors in nucleus accumbens during nicotine withdrawal. Psychopharmacology (Berl). 2011; 213 (4): 735-44.
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008; 154 (2): 384-96.
- Gurfein BT, Stamm AW, Bacchetti P, et al. The calm mouse: an animal model of stress reduction. Mol Med. 2012; 18 (1): 606-17.
- Burgdorf J, Panksepp J. The neurobiology of positive emotions. Neurosci Biobehav Rev. 2006; 30 (2): 173-87.
- Al-Wadei HA, Plummer HK 3rd, Ullah MF, et al. Social stress promotes and gamma-aminobutyric acid inhibits tumor growth in mouse models of non-small cell lung cancer. Cancer Prev Res (Phila). 2012; 5 (2): 189-96.
- Chen H, Liu D, Guo L, et al. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating beta-adrenergic signaling. J Pathol. 2018; 244 (1): 49-60.
- Sloan EK, Priceman SJ, Cox BF, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010; 70 (18): 7042-52.
- Sood AK, Bhatty R, Kamat AA, et al. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006; 12 (2): 369-75.
- Kawai H, Berg DK. Nicotinic acetylcholine receptors containing alpha 7 subunits on rat cortical neurons do not undergo long-lasting inactivation even when up-regulated by chronic nicotine exposure. J Neurochem. 2001; 78 (6): 1367-78.
- Al-Wadei MH, Al-Wadei HA, Schuller HM. Effects of chronic nicotine on the autocrine regulation of pancreatic cancer cells and pancreatic duct epithelial cells by stimulatory and inhibitory neurotransmitters. Carcinogenesis. 2012; 33 (9): 1745-53.
- Muthuswamy R, Okada NJ, Jenkins FJ, et al. Epinephrine promotes COX-2-dependent immune suppression in myeloid cells and cancer tissues. Brain Behav Immun. 2017; 62: 78-86.